Sexual selection in females and the evolution of polyandry
Supplementary material reporting R code
Salomé Fromonteil1†, Lucas
Marie-Orleach2,3†, Lennart Winkler4, Tim
Janicke1,4*
1 CEFE, Univ Montpellier,
CNRS, EPHE, IRD, Montpellier 34090, France.
2 Natural
History Museum, University of Oslo, NO-0318, Oslo, Norway.
3 CNRS, Université de Rennes 1, ECOBIO (Écosystèmes,
biodiversité, évolution) - UMR 6553, F-35000, Rennes, France.
4 Applied Zoology, TU Dresden, Zellescher Weg 20b, 01062
Dresden, Germany.
† Both authors contributed equally to
this work.
* Corresponding author: tim.janicke@cefe.cnrs.fr
Last updated: 2022-11-08
Checks: 7 0
Knit directory: META_SexSelFem/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20211007) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 9e4feda. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: data/.DS_Store
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/index.Rmd) and HTML
(docs/index.html) files. If you’ve configured a remote Git
repository (see ?wflow_git_remote), click on the hyperlinks
in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 9e4feda | Salomé Fromonteil | 2022-11-08 | workflowr::wflow_publish(files = "analysis/index.Rmd") |
| html | 0f4a857 | Salomé Fromonteil | 2022-07-20 | Build site. |
| Rmd | 81dc631 | Salomé Fromonteil | 2022-07-20 | workflowr::wflow_publish(files = "analysis/index.Rmd") |
| Rmd | 540f08c | Salomé Fromonteil | 2022-07-20 | Update for review |
| html | 540f08c | Salomé Fromonteil | 2022-07-20 | Update for review |
| html | be3338c | GitHub | 2021-10-23 | Update index.html |
| html | 81d37fc | Salomé Fromonteil | 2021-10-23 | Build site. |
| Rmd | 4149f69 | Salomé Fromonteil | 2021-10-23 | wflow_publish(files = c("analysis/index.Rmd")) |
| html | f76a4de | Salomé Fromonteil | 2021-10-23 | Build site. |
| Rmd | b901734 | Salomé Fromonteil | 2021-10-23 | wflow_publish(files = c("analysis/index.Rmd")) |
| html | 6134356 | Salomé Fromonteil | 2021-10-23 | Build site. |
| html | 63b9eaa | Salomé Fromonteil | 2021-10-23 | Build site. |
| Rmd | 777f71b | Salomé Fromonteil | 2021-10-23 | wflow_publish(files = c("analysis/index.Rmd")) |
| html | 063fd98 | Salomé Fromonteil | 2021-10-23 | Build site. |
| html | 78ed0b4 | Salomé Fromonteil | 2021-10-23 | Build site. |
| Rmd | 0ded75c | Salomé Fromonteil | 2021-10-23 | wflow_publish(files = c("analysis/index.Rmd")) |
| html | bcbc596 | Salomé Fromonteil | 2021-10-23 | Build site. |
| Rmd | 85df287 | Salomé Fromonteil | 2021-10-23 | wflow_publish(files = c("analysis/index.Rmd")) |
| html | e9f28a6 | Salomé Fromonteil | 2021-10-23 | Build site. |
| Rmd | 6cc53ab | Salomé Fromonteil | 2021-10-23 | wflow_publish(all = TRUE, republish = TRUE) |
| html | 6cc53ab | Salomé Fromonteil | 2021-10-23 | wflow_publish(all = TRUE, republish = TRUE) |
| html | 4faf611 | Salomé Fromonteil | 2021-10-22 | Build site. |
| html | b28958b | Salomé Fromonteil | 2021-10-22 | Start project |
| html | 24b1d38 | GitHub | 2021-10-22 | Add files via upload |
| html | 54bd8aa | salfromo | 2021-10-21 | wflow_git_commit(all = TRUE) |
| html | 018517f | salfromo | 2021-10-21 | Build site. |
| Rmd | 0da8dd8 | Salom<e9> Fromonteil | 2021-10-07 | Start workflowr project. |
# load packages
library(Matrix)
library(metafor)
library(ape)
library(coda)
library(MCMCglmm)
library(ggplot2)
library(cowplot)
library(plyr)
library(dplyr)
library(data.table)
library(ggtree)
library(RColorBrewer)
library(grid)
library(ggnewscale)
library(ggtext)
library(kableExtra)
library(htmlTable)
library(knitr)
library(ggtext)
knitr::opts_chunk$set(warning = FALSE, message = FALSE) # Suppress warnings in the HTML output
knitr::opts_chunk$set(dev = c("png", "pdf")) # saves figures as png and pdf but prints png# Plot aesthetics ####
# Colour palettes
Palette <- colorRampPalette(brewer.pal(n = 9, name = 'BrBG'))
TealPalette <- colorRampPalette(Palette(20)[10:20])
CoffeePalette <- colorRampPalette(rev(Palette(20)[1:10]))
# Plot themes
theme_min <- function(...) { # removes grids and backgrounds in plots
theme(panel.border = element_blank(),
panel.background = element_rect(fill = "transparent", colour = NA),
plot.background = element_rect(fill = "transparent", colour = NA),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
legend.position = "none") +
theme(...)
}
theme_paper <- function(...) { # basic theme function to apply to all plots
theme_min(axis.line.x = element_line(colour = "black", size = 1),
axis.line.y = element_line(colour = "black", size = 1),
axis.text.x = element_text(face = "plain", colour = "black", size = 16, angle = 0),
axis.text.y = element_text(face = "plain", colour = "black", size = 16, angle = 0),
axis.title.x = element_text(face = "plain", size = 16, margin = margin(r = 0, 10, 0, 0)),
axis.title.y = element_text(face = "plain", size = 16, margin = margin(r = 10, 0, 0, 0)),
axis.ticks = element_line(size = 1, colour = "black"),
axis.ticks.length = unit(.3, "cm")) +
theme(...)
}
theme_density <- function(...) { # basic theme + theme specific to density plots
theme_paper(legend.position = c(0.8, 0.975),
legend.title = element_blank(),
legend.text = element_text(colour = "black", size = 16),
legend.key = element_rect(colour = "transparent", fill = "transparent")) +
theme(...)
}
theme_jitter <- function(...) { # density theme + theme specific to jitter plots
theme_density(legend.position = "none",
axis.ticks = element_line(colour = "transparent"),
axis.text.y = element_text(colour = "transparent")) +
theme(...)
}
draw_key_spaced <- function(data, params, size) { # resizes legend keys
rectGrob(width = unit(0.8, "npc"),
height = unit(0.8, "npc"),
gp = gpar(col = data$colour,
fill = alpha(data$fill, data$alpha)))
}
GeomDensity$draw_key <- draw_key_spacedData
To address the question whether sexual selection in females is
widespread, we screened published papers to find female Bateman
gradients (see Figure S5 and Materials and Methods). The dataset
includes 111 effect sizes, extracted from 78 studies and across 72
species.
Source Data Table of effect sizes included in the meta-analysis.
Data <- read.table("./data/Fromonteil_et_al_DATA.txt", fileEncoding = "UTF-8") # the file encoding supports special characters
Tree <- read.tree("./data/Fromonteil_et_al_TREE.txt")
Data %>%
dplyr::select(-c(4, 7, 14)) %>%
mutate_if(is.numeric, round, digits = 3) %>%
kable("html") %>%
kable_paper(full_width = T) %>%
scroll_box(width = "100%", height = "500px") %>%
kable_styling(fixed_thead = T)| Index | Study_ID | Study | Species_Phylo | Class | Polyandry | MatingSystem | StudyType | MS_estimate | Zero_f | beta_ss | r_f | r_VAR_f | N_f |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | Gerlach et al. 2012 | Junco_hyemalis | Aves | 0.439 | low polyandry | Field | gMS | 0 | 0.246 | 0.303 | 0.002 | 436 |
| 2 | 3 | Gopurenko et al 2007 | Ambystoma_texanum | Amphibia | 0.857 | high polyandry | Field | gMS | 1 | 18.342 | 0.817 | 0.003 | 37 |
| 4 | 4 | Gopurenko et al. 2006 | Ambystoma_tigrinum | Amphibia | 0.467 | low polyandry | Field | gMS | 0 | 0.078 | 0.065 | 0.071 | 15 |
| 5 | 9 | Jones et al. 2002 | Taricha_granulosa | Amphibia | 0.548 | high polyandry | Field | gMS | 0 | 22.200 | 0.269 | 0.030 | 30 |
| 6 | 10 | Jones et al. 2004 | Taricha_granulosa | Amphibia | 0.548 | high polyandry | Lab | gMS | 1 | 21.700 | 0.456 | 0.126 | 6 |
| 7 | 10 | Jones et al. 2004 | Taricha_granulosa | Amphibia | 0.548 | high polyandry | Lab | gMS | 1 | -18.700 | -0.206 | 0.183 | 6 |
| 8 | 11 | Jones et al. 2000 | Syngnathus_typhle | Actinopterygii | 0.528 | high polyandry | Lab | gMS | 1 | 24.100 | 0.834 | 0.002 | 62 |
| 9 | 11 | Jones et al. 2000 | Syngnathus_typhle | Actinopterygii | 0.528 | high polyandry | Lab | gMS | 1 | 14.500 | 0.588 | 0.039 | 12 |
| 10 | 14 | Krakauer 2008 | Meleagris_gallopavo | Aves | 0.450 | low polyandry | Field | gMS | 0 | 2.299 | 0.336 | 0.028 | 29 |
| 11 | 16 | Jones et al. 2012 | Spermophilus_columbianus | Mammalia | 0.343 | low polyandry | Field | gMS | 0 | 0.280 | 0.225 | 0.009 | 99 |
| 12 | 17 | Levitan 2008 | Strongylocentrotus_franciscanus | Echinoidea | 0.990 | high polyandry | Field | gMS | 1 | 0.007 | 0.025 | 0.006 | 178 |
| 13 | 17 | Levitan 2008 | Strongylocentrotus_purpuratus | Echinoidea | 0.990 | high polyandry | Field | gMS | 1 | -0.040 | -0.115 | 0.020 | 50 |
| 14 | 19 | Mills et al. 2007 | Myodes_glareolus | Mammalia | 0.353 | low polyandry | Field | gMS | 1 | 0.200 | 0.175 | 0.085 | 12 |
| 15 | 19 | Mills et al. 2007 | Myodes_glareolus | Mammalia | 0.353 | low polyandry | Field | gMS | 1 | -0.250 | -0.180 | 0.134 | 8 |
| 16 | 19 | Mills et al. 2007 | Myodes_glareolus | Mammalia | 0.353 | low polyandry | Field | gMS | 1 | 0.000 | 0.010 | 0.200 | 6 |
| 17 | 20 | Mobley & Jones 2013 | Syngnathus_floridae | Actinopterygii | 0.633 | high polyandry | Field | gMS | 1 | 0.930 | 0.897 | 0.001 | 50 |
| 19 | 21 | Munroe & Koprowski 2011 | Xerospermophilus_tereticaudus | Mammalia | 0.935 | high polyandry | Field | gMS | 0 | 0.810 | 0.883 | 0.002 | 31 |
| 21 | 22 | Moorad et al. 2011 | Homo_sapiens | Mammalia | 0.012 | low polyandry | Field | cMS | 1 | 0.773 | 0.011 | 0.000 | 21865 |
| 22 | 22 | Moorad et al. 2011 | Homo_sapiens | Mammalia | 0.012 | low polyandry | Field | cMS | 1 | 0.332 | 0.008 | 0.000 | 6731 |
| 23 | 23 | Borgerhoff Mulder 2009 | Homo_sapiens | Mammalia | 0.012 | low polyandry | Field | cMS | 1 | 0.196 | 0.052 | 0.007 | 154 |
| 24 | 24 | Croshaw 2010 | Ambystoma_opacum | Amphibia | 0.294 | low polyandry | Lab | gMS | 0 | 11.300 | 0.158 | 0.043 | 23 |
| 25 | 24 | Croshaw 2010 | Ambystoma_opacum | Amphibia | 0.294 | low polyandry | Lab | gMS | 0 | 15.150 | 0.238 | 0.044 | 21 |
| 26 | 27 | Schlicht & Kempenears 2013 | Cyanistes_caeruleus | Aves | 0.470 | low polyandry | Field | gMS | 1 | -0.010 | -0.002 | 0.002 | 447 |
| 27 | 28 | Rodriguez-Munoz et al. 2010 | Gryllus_campestris | Arthropoda | 0.809 | high polyandry | Field | cMS | 1 | 0.840 | 0.133 | 0.018 | 55 |
| 28 | 29 | Fritzsche & Arnqvist 2013 | Megabruchidius_tonkineus | Arthropoda | 0.977 | high polyandry | Lab | cMS | 1 | 0.872 | 0.755 | 0.004 | 50 |
| 29 | 29 | Fritzsche & Arnqvist 2013 | Megabruchidius_dorsalis | Arthropoda | 1.000 | high polyandry | Lab | cMS | 1 | 0.617 | 0.610 | 0.008 | 50 |
| 30 | 29 | Fritzsche & Arnqvist 2013 | Callosobruchus_maculatus | Arthropoda | 1.000 | high polyandry | Lab | cMS | 1 | 0.171 | 0.230 | 0.018 | 50 |
| 31 | 29 | Fritzsche & Arnqvist 2013 | Callosobruchus_chinensis | Arthropoda | 0.680 | high polyandry | Lab | cMS | 1 | 0.096 | 0.195 | 0.019 | 50 |
| 32 | 31 | Anthes et al. 2010 | Biomphalaria_glabrata | Gastropoda | 0.654 | high polyandry | Lab | cMS | 1 | 0.226 | 0.364 | 0.026 | 30 |
| 34 | 32 | Ketterson et al. 1997 | Junco_hyemalis | Aves | 0.439 | low polyandry | Field | gMS | 1 | 2.920 | 0.769 | 0.004 | 45 |
| 35 | 36 | Garcia-Navas et al. 2014 | Cyanistes_caeruleus | Aves | 0.470 | low polyandry | Field | gMS | 0 | 1.117 | 0.345 | 0.012 | 65 |
| 36 | 38 | Gagnon et. al. 2012 | Gerris_gillettei | Arthropoda | 0.776 | high polyandry | Field | gMS | 1 | 1.050 | 0.365 | 0.008 | 97 |
| 37 | 39 | Walker et al. 2014 | Notiomystis_cincta | Aves | 0.836 | high polyandry | Field | gMS | 1 | 0.600 | 0.430 | 0.007 | 91 |
| 39 | 1001 | Bergeron et al. 2012 | Tamias_striatus | Mammalia | 0.650 | high polyandry | Field | gMS | 1 | 1.000 | 0.941 | 0.000 | 145 |
| 41 | 1002 | Courtiol et al. 2012 | Homo_sapiens | Mammalia | 0.012 | low polyandry | Field | cMS | 1 | 0.565 | 0.369 | 0.000 | 1669 |
| 43 | 1004 | Andrade & Kasumovic 2005 | Latrodectus_hasseltii | Arthropoda | 0.667 | high polyandry | Lab | cMS | 1 | 0.475 | 0.394 | 0.006 | 119 |
| 46 | 1005 | Balenger et al. 2009 | Sialia_currucoides | Aves | 0.342 | low polyandry | Field | gMS | 0 | 0.034 | 0.067 | 0.017 | 59 |
| 47 | 1006 | Barreto & Avise 2010 | Pycnogonum_stearnsi | Arthropoda | 0.686 | high polyandry | Field | gMS | 1 | 0.990 | 0.877 | 0.001 | 62 |
| 48 | 1007 | Becher & Magurran 2004 | Poecilia_reticulata | Actinopterygii | 0.710 | high polyandry | Lab | gMS | 1 | 0.807 | 0.704 | 0.003 | 99 |
| 50 | 1008 | Bjork & Pitnick 2006 | Drosophila_lummei | Arthropoda | 0.753 | high polyandry | Lab | cMS | 0 | 0.588 | 0.482 | 0.007 | 85 |
| 51 | 1008 | Bjork & Pitnick 2006 | Drosophila_melanogaster | Arthropoda | 0.759 | high polyandry | Lab | cMS | 0 | 0.042 | 0.079 | 0.007 | 140 |
| 52 | 1008 | Bjork & Pitnick 2006 | Drosophila_bifurca | Arthropoda | 0.326 | low polyandry | Lab | cMS | 0 | 0.237 | 0.201 | 0.008 | 114 |
| 53 | 1008 | Bjork & Pitnick 2006 | Drosophila_virilis | Arthropoda | 0.753 | high polyandry | Lab | cMS | 0 | -0.159 | -0.221 | 0.022 | 42 |
| 54 | 1009 | Collet et al. 2012 | Gallus_gallus | Aves | 0.950 | high polyandry | Lab | cMS | 1 | 0.290 | 0.608 | 0.033 | 13 |
| 55 | 1010 | Fitze & Le Galliard 2011 | Zootoca_vivipara | Reptilia | 0.633 | high polyandry | Field | gMS | 1 | 1.482 | 0.950 | 0.002 | 6 |
| 56 | 1010 | Fitze & Le Galliard 2011 | Zootoca_vivipara | Reptilia | 0.633 | high polyandry | Field | gMS | 1 | 0.947 | 0.890 | 0.009 | 6 |
| 59 | 1011 | Jokela et al. 2010 | Homo_sapiens | Mammalia | 0.012 | low polyandry | Field | cMS | 1 | 0.120 | 0.089 | 0.000 | 4010 |
| 60 | 1013 | Aronsen et al. 2013 | Syngnathus_typhle | Actinopterygii | 0.528 | high polyandry | Lab | gMS | 1 | 0.956 | 0.977 | 0.000 | 50 |
| 61 | 1013 | Aronsen et al. 2013 | Syngnathus_typhle | Actinopterygii | 0.528 | high polyandry | Lab | gMS | 1 | 0.987 | 0.895 | 0.000 | 100 |
| 62 | 1013 | Aronsen et al. 2013 | Syngnathus_typhle | Actinopterygii | 0.528 | high polyandry | Lab | gMS | 1 | 0.855 | 0.729 | 0.002 | 100 |
| 63 | 1013 | Aronsen et al. 2013 | Syngnathus_typhle | Actinopterygii | 0.528 | high polyandry | Lab | gMS | 1 | 1.040 | 0.662 | 0.006 | 50 |
| 64 | 1014 | Rose et al. 2013 | Syngnathus_scovelli | Actinopterygii | 0.615 | high polyandry | Lab | gMS | 1 | 0.915 | 0.893 | 0.001 | 56 |
| 66 | 1017 | Ursprung et al. 2011 | Allobates_femoralis | Amphibia | 0.571 | high polyandry | Field | gMS | 0 | 0.770 | 0.542 | 0.012 | 42 |
| 68 | 2001 | Pélissié et al. 2012 | Physa_acuta | Gastropoda | 0.789 | high polyandry | Lab | cMS | 1 | 0.336 | 0.260 | 0.007 | 120 |
| 71 | 2002 | Pongratz & Michiels 2003 | Schmidtea_polychroa | Plathelminthes | 0.918 | high polyandry | Lab | gMS | 1 | 0.965 | 0.779 | 0.003 | 54 |
| 73 | 2003 | Poesel et al. 2011 | Zonotrichia_leucophrys | Aves | 0.346 | low polyandry | Field | gMS | 0 | 0.151 | 0.196 | 0.018 | 52 |
| 74 | 2004 | Prosser et al. 2002 | Nerodia_sipedon | Reptilia | 0.556 | high polyandry | Field | gMS | 0 | 1.789 | 0.381 | 0.017 | 45 |
| 76 | 2005 | Rios-Cardenas 2005 | Lepomis_gibbosus | Actinopterygii | 0.244 | low polyandry | Field | gMS | 0 | 64.170 | 0.031 | 0.008 | 123 |
| 77 | 2006 | Schulte-Hostedde et al. 2004 | Tamias_amoenus | Mammalia | 0.595 | high polyandry | Field | gMS | 1 | 0.857 | 0.870 | 0.002 | 37 |
| 79 | 2008 | Tatarenkov et al. 2008 | Xiphophorus_helleri | Actinopterygii | 0.638 | high polyandry | Field | gMS | 0 | 0.407 | 0.231 | 0.022 | 42 |
| 81 | 2008 | Tatarenkov et al. 2008 | Xiphophorus_helleri | Actinopterygii | 0.638 | high polyandry | Field | gMS | 0 | 0.234 | 0.152 | 0.037 | 27 |
| 83 | 2009 | Whittingham & Dunn 2005 | Geothlypis_trichas | Aves | 0.654 | high polyandry | Field | gMS | 0 | 0.757 | 0.323 | 0.045 | 19 |
| 84 | 2010 | Williams & DeWoody 2009 | Ambystoma_tigrinum | Amphibia | 0.467 | low polyandry | Field | gMS | 1 | 1.036 | 0.751 | 0.004 | 45 |
| 85 | 2011 | Woolfenden et al. 2002 | Molothrus_ater | Aves | 0.455 | low polyandry | Field | gMS | 1 | 1.910 | 0.700 | 0.002 | 142 |
| 86 | 2014 | Serbezov et al. 2010 | Salmo_trutta | Actinopterygii | 0.680 | high polyandry | Field | gMS | 1 | 1.026 | 0.970 | 0.000 | 116 |
| 88 | 2014 | Serbezov et al. 2010 | Salmo_trutta | Actinopterygii | 0.680 | high polyandry | Field | gMS | 1 | 0.967 | 0.718 | 0.002 | 121 |
| 89 | 2014 | Serbezov et al. 2010 | Salmo_trutta | Actinopterygii | 0.680 | high polyandry | Field | gMS | 1 | 1.117 | 0.576 | 0.004 | 107 |
| 90 | 2015 | Whittingham & Lifjeld 1995 | Delichon_urbica | Aves | 0.235 | low polyandry | Field | gMS | 0 | -0.057 | -0.092 | 0.076 | 14 |
| 91 | 2016 | Broquet et al. 2009 | Hyla_arborea | Amphibia | 0.158 | low polyandry | Field | gMS | 0 | 0.438 | 0.159 | 0.053 | 19 |
| 93 | 2020 | Byers et al. 2004 | Dendroica_pensylvanica | Aves | 0.606 | high polyandry | Field | gMS | 1 | 0.686 | 0.523 | 0.015 | 37 |
| 94 | 3001 | Buerkli & Jokela 2017 | Radix_balthica | Gastropoda | 0.500 | high polyandry | Field | gMS | 0 | -0.010 | -0.173 | 0.016 | 60 |
| 96 | 3003 | Cattelan et al. 2020 | Poecilia_reticulata | Actinopterygii | 0.710 | high polyandry | Lab | cMS | 1 | 0.175 | 0.121 | 0.016 | 60 |
| 98 | 3003 | Cattelan et al. 2020 | Poecilia_reticulata | Actinopterygii | 0.710 | high polyandry | Lab | cMS | 1 | 0.198 | 0.135 | 0.019 | 53 |
| 100 | 3004 | Grunst et al. 2019 | Zonotrichia_albicollis | Aves | 0.638 | high polyandry | Field | gMS | 1 | 0.193 | 0.180 | 0.004 | 222 |
| 101 | 3005 | Hoffer et al. 2017 | Lymnaea_stagnalis | Gastropoda | 0.588 | high polyandry | Lab | cMS | 1 | -0.083 | -0.082 | 0.047 | 22 |
| 102 | 3006 | Johannesson et al. 2016 | Littorina_saxatilis | Gastropoda | 0.680 | high polyandry | Lab | gMS | 0 | 0.055 | 0.071 | 0.041 | 25 |
| 105 | 3007 | Devost & Turgeon 2016 | Gerris_buenoi | Arthropoda | 1.000 | high polyandry | Lab | cMS | 0 | -0.416 | -0.203 | 0.027 | 35 |
| 106 | 3008 | Scheepers & Gouws 2019 | Clinus_cottoides | Actinopterygii | 0.826 | high polyandry | Field | gMS | 0 | 0.440 | 0.265 | 0.039 | 23 |
| 107 | 3009 | Wang et al. 2020 | Acanthopagrus_schlegelii | Actinopterygii | 0.920 | high polyandry | Lab | gMS | 0 | 1.746 | 0.933 | 0.001 | 25 |
| 108 | 3010 | Louder et al. 2019 | Molothrus_ater | Aves | 0.455 | low polyandry | Field | gMS | 1 | 0.480 | 0.177 | 0.009 | 100 |
| 110 | 3011 | Janicke et al. 2015 | Physa_acuta | Gastropoda | 0.789 | high polyandry | Lab | cMS | 1 | 0.543 | 0.534 | 0.013 | 39 |
| 112 | 3011 | Janicke et al. 2015 | Physa_acuta | Gastropoda | 0.789 | high polyandry | Lab | cMS | 1 | 0.007 | 0.005 | 0.026 | 39 |
| 114 | 3012 | Levine et al. 2015 | Agkistrodon_contortrix | Reptilia | 0.520 | high polyandry | Field | gMS | 0 | 0.070 | 0.032 | 0.055 | 19 |
| 115 | 3013 | Borgerhoff Mulder & Ross 2019 | Homo_sapiens | Mammalia | 0.012 | low polyandry | Field | cMS | 1 | 0.100 | 0.163 | 0.005 | 176 |
| 116 | 3014 | Bolopo et al. 2016 | Clamator_glandarius | Aves | 0.697 | high polyandry | Field | gMS | 0 | 1.080 | 0.727 | 0.003 | 65 |
| 117 | 3015 | Glaudas et al. 2020 | Bitis_arietans | Reptilia | 0.654 | high polyandry | Field | gMS | 0 | 2.480 | 0.386 | 0.045 | 17 |
| 118 | 3016 | Saunders & Shuster 2019 | Paracerceis_sculpta | Arthropoda | 0.789 | high polyandry | Lab | cMS | 0 | 1.780 | 0.195 | 0.185 | 6 |
| 119 | 3017 | Paczolt et al. 2015 | Xiphophorus_birchmanni | Actinopterygii | 0.839 | high polyandry | Field | gMS | 0 | 0.702 | 0.613 | 0.013 | 31 |
| 120 | 3017 | Paczolt et al. 2015 | Xiphophorus_birchmanni | Actinopterygii | 0.839 | high polyandry | Field | gMS | 0 | 0.987 | 0.551 | 0.044 | 12 |
| 121 | 3018 | Mangold et al. 2015 | Hyalinobatrachium_valerioi | Amphibia | 0.736 | high polyandry | Field | gMS | 1 | 0.940 | 0.895 | 0.001 | 77 |
| 123 | 3019 | Marie-Orleach et al. 2016 | Macrostomum_lignano | Plathelminthes | 1.000 | high polyandry | Lab | cMS | 1 | 0.290 | 0.097 | 0.019 | 52 |
| 124 | 3020 | Kretzschmar et al. 2019 | Ceratotherium_simum | Mammalia | 0.697 | high polyandry | Field | gMS | 0 | 1.059 | 0.830 | 0.003 | 33 |
| 125 | 3021 | Morimoto et al. 2016 | Drosophila_melanogaster | Arthropoda | 0.759 | high polyandry | Lab | cMS | 0 | 0.020 | 0.010 | 0.018 | 58 |
| 126 | 3022 | Turnell and Shaw 2015 | Laupala_cerasina | Arthropoda | 1.000 | high polyandry | Lab | cMS | 0 | 0.197 | 0.292 | 0.042 | 21 |
| 127 | 3023 | Fuxjäger et al. 2019 | Gasterosteus_aculeatus | Actinopterygii | 0.950 | high polyandry | Lab | gMS | 1 | 0.954 | 0.791 | 0.004 | 36 |
| 128 | 3023 | Fuxjäger et al. 2019 | Gasterosteus_aculeatus | Actinopterygii | 0.950 | high polyandry | Lab | gMS | 1 | 0.922 | 0.686 | 0.008 | 36 |
| 131 | 3024 | McCullough et al. 2018 | Onthophagus_taurus | Arthropoda | 0.789 | high polyandry | Lab | gMS | 0 | 0.994 | 0.772 | 0.003 | 50 |
| 132 | 3024 | McCullough et al. 2018 | Onthophagus_taurus | Arthropoda | 0.789 | high polyandry | Lab | gMS | 0 | 0.693 | 0.636 | 0.004 | 89 |
| 133 | 3024 | McCullough et al. 2018 | Onthophagus_taurus | Arthropoda | 0.789 | high polyandry | Lab | gMS | 0 | 1.301 | 0.780 | 0.007 | 22 |
| 134 | 3025 | Wells et al. 2017 | Callospermophilus_lateralis | Mammalia | 0.630 | high polyandry | Field | gMS | 0 | 0.460 | 0.224 | 0.038 | 25 |
| 137 | 3028 | Skjærvø & Røskaft 2015 | Homo_sapiens | Mammalia | 0.012 | low polyandry | Field | cMS | 0 | 0.160 | 0.144 | 0.001 | 753 |
| 138 | 3028 | Skjærvø & Røskaft 2015 | Homo_sapiens | Mammalia | 0.012 | low polyandry | Field | cMS | 0 | 0.100 | 0.059 | 0.002 | 443 |
| 139 | 3028 | Skjærvø & Røskaft 2015 | Homo_sapiens | Mammalia | 0.012 | low polyandry | Field | cMS | 0 | 0.020 | 0.017 | 0.003 | 395 |
| 140 | 3028 | Skjærvø & Røskaft 2015 | Homo_sapiens | Mammalia | 0.012 | low polyandry | Field | cMS | 0 | -0.020 | -0.012 | 0.005 | 205 |
| 141 | 3029 | Levine et al. 2020 | Crotalus_atrox | Reptilia | 0.400 | low polyandry | Field | gMS | 0 | 0.151 | 0.099 | 0.058 | 18 |
| 142 | 3030 | Fromonteil et al. 2022 | Acanthoscelides_obtectus | Arthropoda | 0.551 | high polyandry | Lab | cMS | 1 | 0.489 | 0.390 | 0.015 | 50 |
| 144 | 3030 | Fromonteil et al. 2022 | Acanthoscelides_obtectus | Arthropoda | 0.551 | high polyandry | Lab | cMS | 1 | 0.910 | 0.525 | 0.011 | 50 |
| 146 | 3030 | Fromonteil et al. 2022 | Acanthoscelides_obtectus | Arthropoda | 0.551 | high polyandry | Lab | cMS | 0 | 0.688 | 0.308 | 0.015 | 55 |
| 147 | 3030 | Fromonteil et al. 2022 | Acanthoscelides_obtectus | Arthropoda | 0.551 | high polyandry | Lab | cMS | 1 | 0.025 | 0.020 | 0.019 | 55 |
| 149 | 3031 | LaBrecque et al. 2014 | Hyperprosopon_anale | Actinopterygii | 1.000 | high polyandry | Field | gMS | 0 | 0.330 | 0.512 | 0.024 | 24 |
| 150 | 3031 | LaBrecque et al. 2014 | Cymatogaster_aggregata | Actinopterygii | 0.870 | high polyandry | Field | gMS | 0 | 0.468 | 0.485 | 0.027 | 23 |
| 151 | 3032 | Dekker et al. 2020 | Poecilia_gillii | Actinopterygii | 0.710 | high polyandry | Field | gMS | 0 | 0.770 | 0.629 | 0.012 | 31 |
| 152 | 3033 | Gauthey et al. 2017 | Salmo_trutta | Actinopterygii | 0.545 | high polyandry | Field | gMS | 1 | 1.027 | 0.606 | 0.013 | 32 |
| 153 | 3034 | Gao et al. 2020 | Heliothis_virescens | Arthropoda | 0.655 | high polyandry | Lab | cMS | 1 | 0.735 | 0.539 | 0.004 | 113 |
| 155 | 3035 | Greenway et al. 2021 | Anasa_tristis | Arthropoda | 0.950 | high polyandry | Lab | cMS | 1 | 0.500 | 0.362 | 0.010 | 80 |
| 157 | 3036 | Winkler et al. 2022 | Tribolium_castaneum | Arthropoda | 0.662 | high polyandry | Lab | cMS | 0 | 0.874 | 0.667 | 0.007 | 48 |
| 158 | 3036 | Winkler et al. 2022 | Tribolium_castaneum | Arthropoda | 0.662 | high polyandry | Lab | cMS | 0 | 0.686 | 0.339 | 0.014 | 59 |
| 159 | 3036 | Winkler et al. 2022 | Tribolium_castaneum | Arthropoda | 0.662 | high polyandry | Lab | cMS | 0 | 0.925 | 0.541 | 0.009 | 56 |
| 160 | 3036 | Winkler et al. 2022 | Tribolium_castaneum | Arthropoda | 0.662 | high polyandry | Lab | cMS | 0 | 0.718 | 0.487 | 0.012 | 51 |
| 165 | 3037 | Hargrove et al. 2020 | Oncorhynchus_clarkii | Actinopterygii | 0.552 | high polyandry | Field | gMS | 1 | NA | 0.280 | 0.001 | 776 |
Index: Individual number given to each effect
size (k = 120).
Study_ID: Identification number given to each study
included in the meta-analysis.
Study: Authors and year of the published study.
Species_Phylo: Species (n = 77).
Polyandry: Level of polyandry of the species, bound
between 0 (exclusively monogamous) and 1 (exclusively
polyandrous).
MatingSystem: Mating system of female individuals.
Either with a low or a high polyandry.
StudyType: Field study or laboratory study.
MS_estimate: Mating success can be estimated either by
using the number of genetic mating partners (gMS) or copulatory
mating success (cMS).
Zero_f: Whether this effect size was calculated on a
population which includes (1) or excludes (0) individuals with zero
mating success.
beta_ss: Female Bateman gradients, as reported in the
published studies. Only some Bateman gradients are standardized.
r_f: Pearson correlation coefficients converted from
female Bateman gradients.
r_VAR_f: Variance of the effect size
(r).
N_f: Sample size of the study.
As only some of the extracted Bateman gradients were computed on
relativised data, we converted all obtained slopes into Pearson
correlation coefficients (r). By running linear regression
models between the actual Bateman Gradient and the correlation
coefficients, we can check whether r is a reliable estimate for
the benefit of mating.
# Relationship between r and betaSS ####
Data_stand <- subset(Data, beta_ss_stand == "1")
LM_beta_f_r <- lm(Data_stand$beta_ss ~ Data_stand$r_f)
Scatter_r_versus_betaSS <- ggplot(data = Data_stand,
mapping = aes(x = r_f, y = beta_ss)) +
geom_hline(yintercept = 0, linetype = "longdash", color = "black", size = 0.5) +
geom_vline(xintercept = 0, linetype = "longdash", color = "black", size = 0.5) +
labs(title = "", x = "*r*", y = expression(italic(beta[ss]))) +
scale_y_continuous(limits = c(-0.6, 1.4), breaks = c(-0.6, 0, 0.6, 1.2)) +
scale_x_continuous(limits = c(-0.3, 1.0), breaks = c(0, 0.5, 1.0)) +
geom_smooth(method = lm, se = TRUE, linetype = "longdash", color = "#008080", fill = "#B2D8D8", size = 0.8) +
geom_point(shape = 21, size = 5, fill = "grey", alpha = .6) +
theme_paper(axis.title.x = ggtext::element_markdown()) +
annotate("text", x = 0.8, y = -0.3, label = paste0("R^2 == ", round(summary(LM_beta_f_r)$r.squared, 2)), parse = TRUE, size = 7)
Scatter_r_versus_betaSS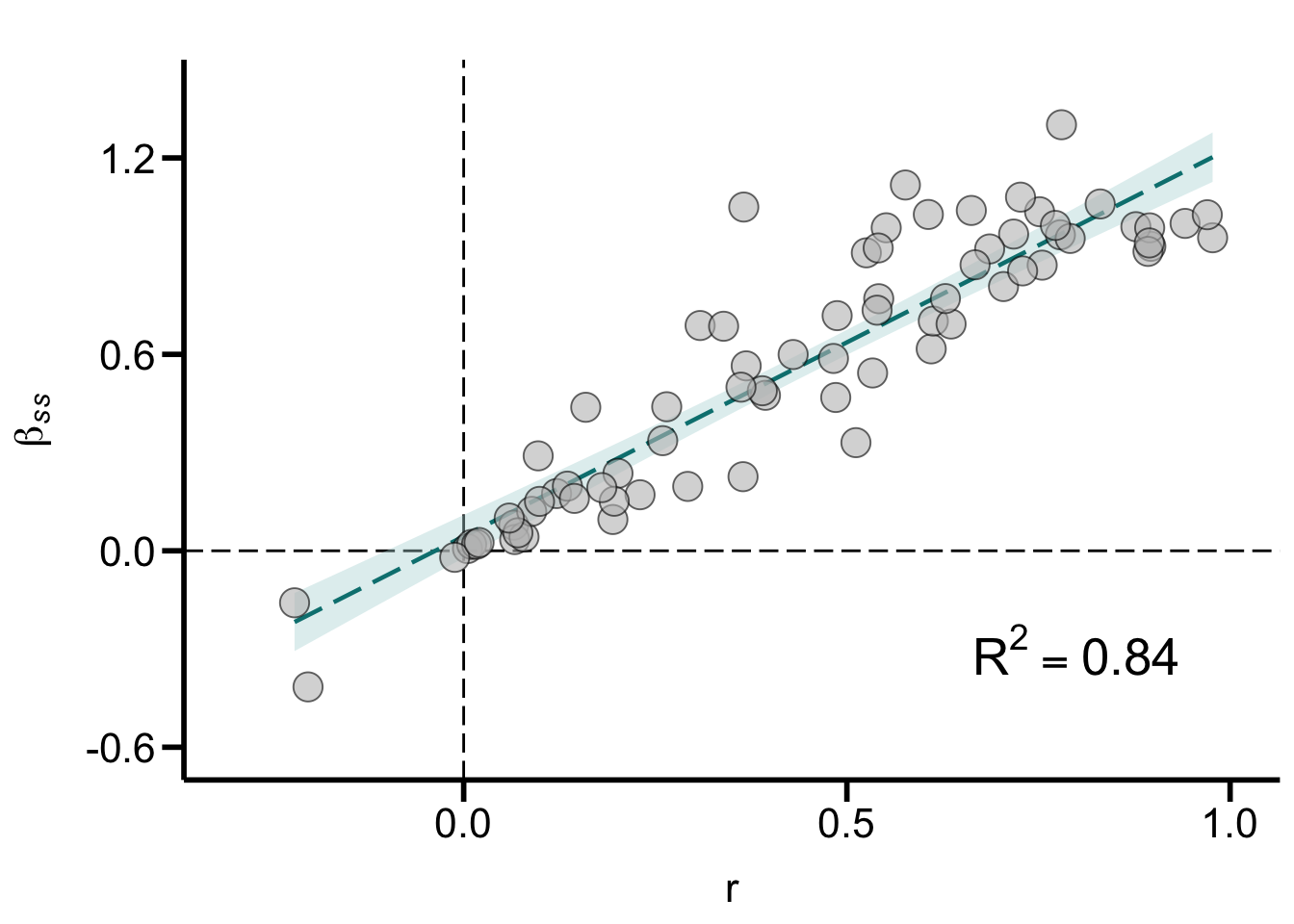
Figure S5A. Relationship between effect size r (correlation coefficient) and the actual Bateman gradient (\(\beta\)ss). Graph shows a subset of data for which standardized Bateman gradients were available (N = 74). Dashed line shows the linear regression fit with shaded area indicating the 95% confidence intervals.
Phylogeny
We reconstructed the phylogeny using divergence times extracted from
the TimeTree database (http://www.timetree.org/, (Kumar
et al. 2017)) and by using the ggtree package (Yu et al. 2017, 2018; Yu 2020).
MetaData_Species <- unique(Data$Species_Phylo)
theTree <- drop.tip(Tree, Tree$tip.label[-na.omit(match(MetaData_Species, Tree$tip.label))])
theTree$tip.label <- gsub("_", " ", theTree$tip.label)
# Phylogenetic tree ####
Phylo <- ggtree(theTree, size = 0.6) +
ylim(-2, 79) +
xlim(-20, 650) +
annotate("rect", xmin = 368, xmax = 570, ymin = 77.5, ymax = 75.5, fill = "#8C510A", alpha = .8) +
annotate("rect", xmin = 368, xmax = 570, ymin = 75.5, ymax = 70.5, fill = "#BF812D", alpha = .8) +
annotate("rect", xmin = 368, xmax = 570, ymin = 70.5, ymax = 50.5, fill = "#DFC27D", alpha = .8) +
annotate("rect", xmin = 368, xmax = 570, ymin = 50.5, ymax = 48.5, fill = "#F6E8C3", alpha = .8) +
annotate("rect", xmin = 368, xmax = 570, ymin = 48.5, ymax = 33.5, fill = "#F5F5F5", alpha = .8) +
annotate("rect", xmin = 368, xmax = 570, ymin = 33.5, ymax = 26.5, fill = "#C7EAE5", alpha = .8) +
annotate("rect", xmin = 368, xmax = 570, ymin = 26.5, ymax = 13.5, fill = "#80CDC1", alpha = .8) +
annotate("rect", xmin = 368, xmax = 570, ymin = 13.5, ymax = 8.5, fill = "#35978F", alpha = .8) +
annotate("rect", xmin = 368, xmax = 570, ymin = 8.5, ymax = 0.0, fill = "#01665E", alpha = .8) +
geom_tiplab(size = 3, color = "black", hjust = 0.0) +
theme_tree()
Phylo <- rotate(Phylo, 78) %>% rotate(82) %>% rotate(84) %>% rotate(96) %>% rotate(100)
# Number of effect sizes/class ####
Data_r_f <- as.data.frame(aggregate(r_f ~ Class + Species_Phylo + Index, data = Data, FUN = mean, na.rm = TRUE))
Data_r_by_Species <- as.data.frame(aggregate(r_f ~ Data_r_f$Class + Data_r_f$Species, data = Data_r_f, FUN = length))
colnames(Data_r_by_Species) <- c("Class", "Species", "N_f_r")
Data_r_by_Class_ES <- as.data.frame(aggregate(N_f_r ~ Class, data = Data_r_by_Species, FUN = sum))
Data_r_by_Class_ES_Species <- as.data.frame(aggregate(N_f_r ~ Class, data = Data_r_by_Species, FUN = length))
Data_r_by_Class_ES$N_species <- Data_r_by_Class_ES_Species$N_f_r
Data_r_by_Class_ES$order <- c(5, 6, 3, 7, 4, 2, 9, 8, 1)
rownames(Data_r_by_Class_ES) <- 1:nrow(Data_r_by_Class_ES)
ES_Sum <- sum(Data_r_by_Class_ES$N_f_r)
DChart_N_ES <- ggplot(data = Data_r_by_Class_ES,
mapping = aes(x = 1, y = N_f_r, fill = as.factor(order))) +
scale_fill_manual(values = Palette(9)) +
geom_bar(stat = "identity") +
xlim(0, 1.5) +
coord_polar(theta = "y", direction = -1) + # polar coordinate system
labs(x = NULL, y = NULL) +
labs(fill = "") +
theme_min(plot.title = element_text(face = "bold", family = "sans", size = 4),
plot.margin = margin(l = -0.5, unit = "cm"),
legend.text = element_text(size = 10),
axis.ticks = element_blank(),
axis.text = element_blank(),
axis.title = element_blank()) +
annotate("text", x = 0, y = 1, label = ES_Sum, size = 5, fontface = "bold", angle = 0)
# Number of species/class ####
Species_Sum <- sum(Data_r_by_Class_ES$N_species)
DChart_N_Species <- ggplot(data = Data_r_by_Class_ES,
mapping = aes(x = 1, y = N_species, fill = as.factor(order))) +
scale_fill_manual(values = Palette(9)) +
geom_bar(stat = "identity") +
xlim(0, 1.5) +
coord_polar(theta = "y", direction = -1) + # polar coordinate system
labs(x = NULL, y = NULL) +
labs(fill = "") +
theme_min(plot.title = element_text(face = "bold", family = "sans", size = 4),
plot.margin = margin(l = -0.5, unit = "cm"),
legend.text = element_text(size = 10),
axis.ticks = element_blank(),
axis.text = element_blank(),
axis.title = element_blank()) +
annotate("text", x = 0, y = 1, label = Species_Sum, size = 5, fontface = "bold", angle = 0)
Figure_Donut_ALL <- plot_grid(DChart_N_ES, DChart_N_Species,
label_size = 15,
hjust = 0,
vjust = 0,
align = "hv",
ncol = 2)
# Combine
ggdraw(Phylo) + draw_plot(Figure_Donut_ALL, x = - 0.33, y = -0.34, scale = 0.3) +
annotate("text", x = 0.085, y = 0.08, label = "Sample\nsize", size = 5, angle = 0, lineheight = 1, hjust = 0.5) +
annotate("text", x = 0.245, y = 0.08, label = "Species\nnumber", size = 5, angle = 0, lineheight = 1, hjust = 0.5)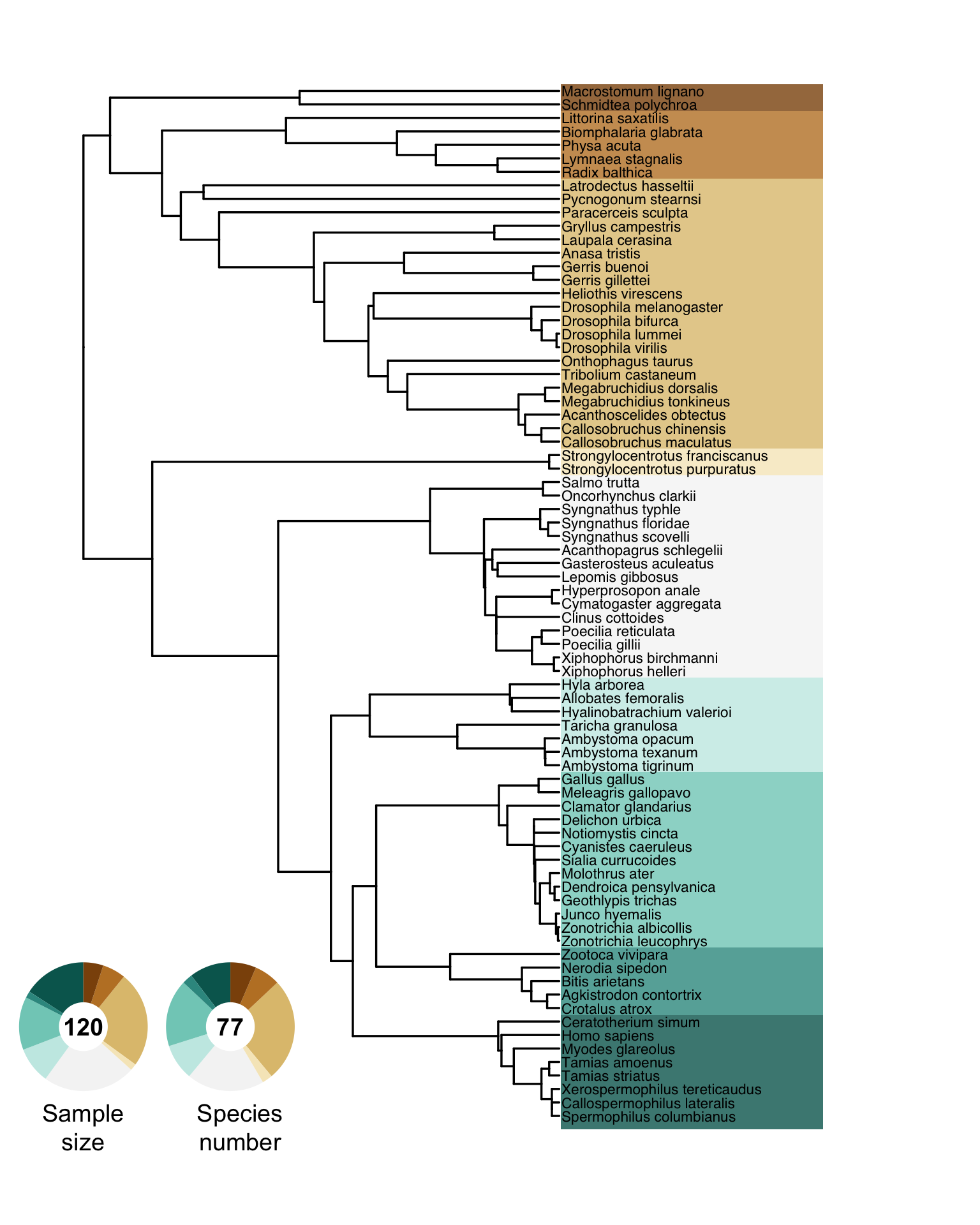
Figure S4. Phylogenetic tree of all sampled species. Doughnut charts show the relative fraction of the sampled effect sizes (i.e., number of Bateman gradients) and the number of species.
Forest plot
Data$r_SE <- sqrt(Data$r_VAR_f) # standard error
Data$l95CI_r_f <- Data$r_f - ((Data$r_SE)*1.96) # 95% confidence interval - lower
Data$u95CI_r_f <- Data$r_f + ((Data$r_SE)*1.96) # 95% confidence interval - upper
ForestData <- Data[order(Data$Phylo_ID), ]
ForestData$Phylo_ID_red <- seq.int(nrow(ForestData))
ForestPlot <- ggplot() +
geom_point(data = ForestData,
mapping = aes(x = r_f, y = Phylo_ID_red),
shape = 23, size = 2, colour = "black") +
geom_errorbarh(data = ForestData,
mapping = aes(y = Phylo_ID_red, xmin = l95CI_r_f, xmax = u95CI_r_f),
size = 0.5, colour = "black", height = 0.0) +
annotate("rect", xmin = 1.2, xmax = 2.2, ymin = 120.5, ymax = 118.5, fill = "#8C510A", alpha = .8) +
annotate("rect", xmin = 1.2, xmax = 2.2, ymin = 118.5, ymax = 111.5, fill = "#BF812D", alpha = .8) +
annotate("rect", xmin = 1.2, xmax = 2.2, ymin = 111.5, ymax = 82.5, fill = "#DFC27D", alpha = .8) +
annotate("rect", xmin = 1.2, xmax = 2.2, ymin = 82.5, ymax = 80.5, fill = "#F6E8C3", alpha = .8) +
annotate("rect", xmin = 1.2, xmax = 2.2, ymin = 80.5, ymax = 52.5, fill = "#F5F5F5", alpha = .8) +
annotate("rect", xmin = 1.2, xmax = 2.2, ymin = 52.5, ymax = 41.5, fill = "#C7EAE5", alpha = .8) +
annotate("rect", xmin = 1.2, xmax = 2.2, ymin = 41.5, ymax = 25.5, fill = "#80CDC1", alpha = .8) +
annotate("rect", xmin = 1.2, xmax = 2.2, ymin = 25.5, ymax = 19.5, fill = "#35978F", alpha = .8) +
annotate("rect", xmin = 1.2, xmax = 2.2, ymin = 19.5, ymax = 0.0, fill = "#01665E", alpha = .8) +
geom_text(data = ForestData,
mapping = aes(label = Study, x = 2.2, y = Phylo_ID_red),
colour = "black", size = 2, angle = 0, check_overlap = FALSE, hjust = "right") +
geom_vline(xintercept = 0, linetype = "dashed", colour = "black", size = 1)+
scale_x_continuous(limits = c(-1.2, 2.2), breaks = c(-0.5, 0, 0.5, 1), expand = c(0 ,0)) +
labs(title = "", x = "Female Bateman gradient (*r*)", y = "Study ID") +
theme_paper(axis.title.x = element_text(size = 18),
axis.title.y = element_text(size = 18)) +
theme(axis.title.x = element_markdown())
ForestPlot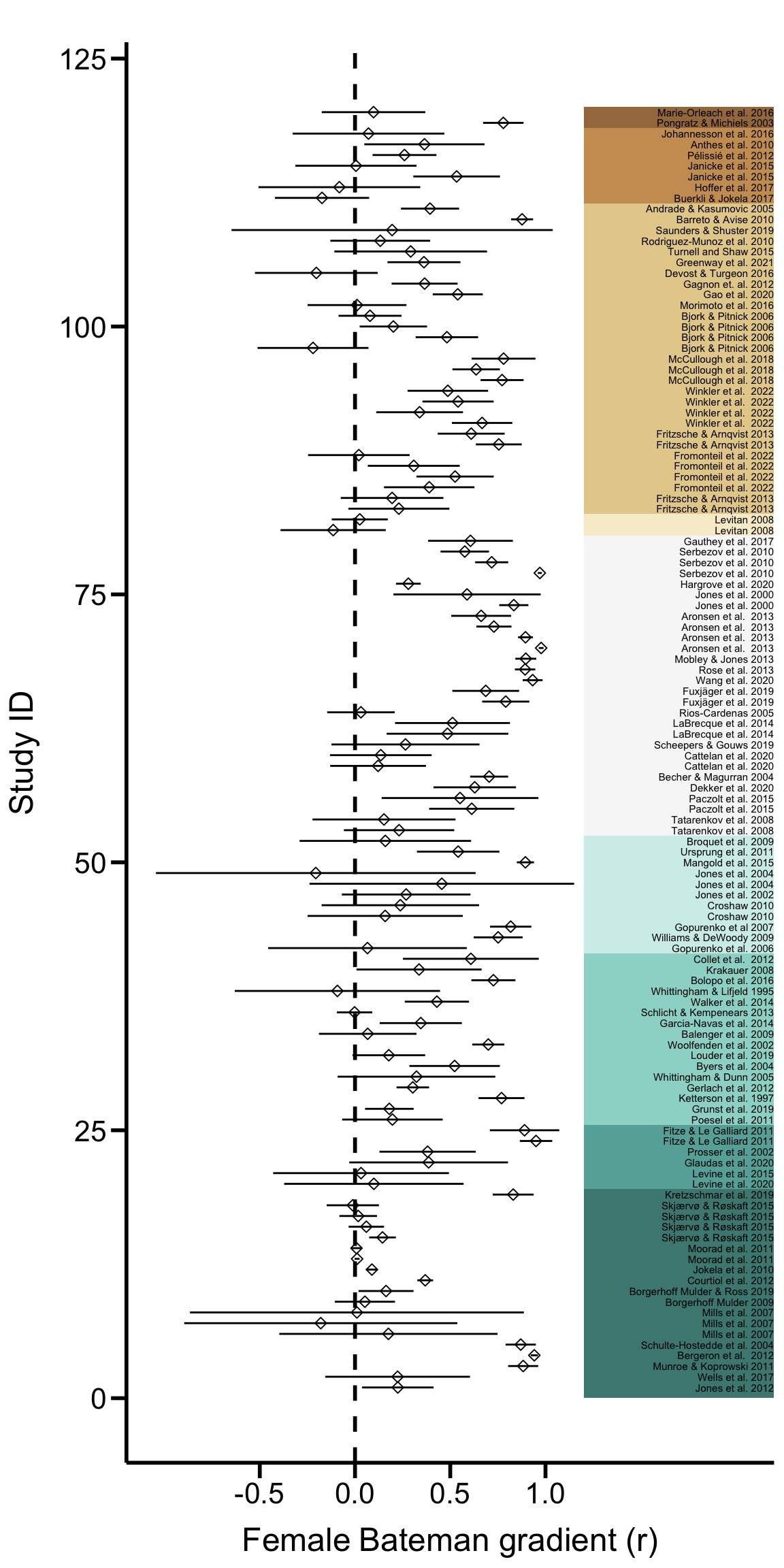
Figure 2. Forest plot. Graphs shows all sampled
effect sizes (Pearson correlation coefficient of Bateman gradients) with
95% confidence limits in phylogenetic order.
Bayesian Meta-Analysis
We ran General Linear Mixed-Effects Models (GLMMs) with the
MCMCglmm::MCMCglmm function (Hadfield 2010) to provide a global test for
sexual selection in females and to explore determinants of the
inter-study variation. We used uninformative priors (V = 1,
nu = 0.002) and an effective sample size of 10,000 (number of
iterations = 4,400,000, burn-in = 400,000, thinning interval = 400).
pr <- list(R = list(V = 1, nu = 0.002), G = list(G1 = list(V = 1, nu = 0.002),
G2 = list(V = 1, nu = 0.002)))
pr2 <- list(R = list(V = 1, nu = 0.002), G = list(G1 = list(V = 1, nu = 0.002),
G2 = list(V = 1, nu = 0.002),
G3 = list(V = 1, nu = 0.002)))
BURNIN = 400000
NITT = 4400000
THIN = 400GLMMs were run with the effect size (r) defined as the response variable weighted by the inverse of its sampling size and included study identifier and observation identifier as a random term.
Global effect size: traditionnal and phylogenetic approach
First, we quantified global effect sizes both without (i.e., ‘non-phylogenetic’ GLMMs) and with adding the phylogenetic correlation matrix as an additional random term (‘phylogenetic’ GLMMs).
Note that all plots and tables presented below might very slightly vary from the ones presented in the manuscript, as the Bayesian approach yields different outputs every time the models are run. However, this do not lead to a qualitative change of results.
names(Data)[names(Data) == "Species_Phylo"] <- "animal" # MCMCglmmm requires the species names to be in a column named 'animal'
# models ####
Model_ALL_traditional <- MCMCglmm(r_f~1,
random = ~Study_ID + Index,
mev = Data$r_VAR_f,
data = Data,
prior = pr, pr = TRUE, verbose = FALSE,
burnin = BURNIN, nitt = NITT, thin = THIN)
Model_ALL_phylo <- MCMCglmm(r_f~1,
random = ~animal + Study_ID + Index,
mev = Data$r_VAR_f,
pedigree = Tree,
data = Data,
prior = pr2, pr = TRUE, verbose = FALSE,
burnin = BURNIN, nitt = NITT, thin = THIN)
# Raincloud plot ####
# density
Data_trad <- as.data.frame(Model_ALL_traditional$Sol[,"(Intercept)"])
Data_trad$Approach <- as.factor(rep("Non-phylogenetic", nrow(Data_trad)))
colnames(Data_trad) <- c("ES", "Approach")
Data_phylo <- as.data.frame(Model_ALL_phylo$Sol[,"(Intercept)"])
Data_phylo$Approach <- as.factor(rep("Phylogenetic", nrow(Data_phylo)))
colnames(Data_phylo) <- c("ES", "Approach")
Data_BothApproaches_ALL <- as.data.frame(rbind(Data_trad, Data_phylo))
Plot_AllApproaches1 <- ggplot() +
geom_density(data = subset(Data_BothApproaches_ALL, Approach == "Non-phylogenetic"),
mapping = aes(x = ES, y = ..density.., fill = Approach),
alpha = 0.7) +
geom_density(data = subset(Data_BothApproaches_ALL, Approach == "Phylogenetic"),
mapping = aes(x = ES, y = ..density.., fill = Approach),
alpha = 0.7) +
scale_colour_manual(values = c(CoffeePalette(20)[5], CoffeePalette(20)[17])) +
scale_fill_manual(values = c(CoffeePalette(20)[5], CoffeePalette(20)[17])) +
geom_vline(xintercept = 0, linetype = "longdash", colour = "black", size = 0.7) +
labs(title = "", y = "Density", x = "Female Bateman gradient (*r*)") +
scale_x_continuous(limits = c(-0.4, 1.1), breaks = c(-0.4, 0, 0.4, 0.8)) +
scale_y_continuous(limits = c(-3, max(density(Data_trad$ES)$y)),
breaks = c(0, 2, 4, 6, 8, 10)) +
theme_density(axis.title.x = element_markdown()) +
theme(legend.spacing = unit(5.0, "in"))
# jitter
CI <- HPDinterval(mcmc(Model_ALL_phylo$Sol[,"(Intercept)"]))
aggdata_BothApproches <- data.frame("Approach" = c("A", "B", "C", "D", "E"),
"Mean" = c(mean(Data_phylo$ES), 2, 3, 4, 5),
"l025" = c(CI[1], 2, 3, 4, 5),
"u975" = c(CI[2], 2, 3, 4, 5))
aggdata_BothApproches$Approach <- as.factor(aggdata_BothApproches$Approach)
Data$Jitter2 <- rep("A", nrow(Data))
rm(CI)
Plot_AllApproaches2 <- ggplot() +
geom_pointrange(data = aggdata_BothApproches,
mapping = aes(x = Mean,
y = Approach,
xmin = l025,
xmax = u975,
colour = Approach),
shape = 23,
size = 1.5,
fatten = 5,
stroke = 1.75,
fill = "white") +
scale_colour_manual(values = c(CoffeePalette(20)[18], "black", "black", "black", "black")) +
new_scale_colour() +
geom_jitter(Data,
mapping = aes(x = r_f,
y = Jitter2,
colour = Jitter2,
fill = Jitter2),
size = log(1 / Data$r_VAR_f),
alpha = 0.4,
position = position_jitter(height = .35)) +
scale_fill_manual(values = CoffeePalette(20)[19]) +
scale_colour_manual(values = CoffeePalette(20)[19]) +
new_scale_colour() +
geom_point(data = aggdata_BothApproches,
mapping = aes(x = Mean,
y = Approach,
colour = Approach),
shape = 23,
size = 6.5,
stroke = 1.75,
fill = alpha("white", .75)) +
geom_vline(xintercept = 0, linetype = "longdash", colour = "black", size = 0.7) +
scale_colour_manual(values = c(CoffeePalette(20)[18], "black", "black", "black", "black")) +
guides("fill" = "none", "colour" = "none") +
scale_x_continuous(limits = c(-0.4, 1.1), breaks = c(-0.4, 0, 0.4, 0.8)) +
scale_y_discrete(labels = c("0", "1", "2", "3", "10")) +
labs(title = "", y = "Density", x = "Female Bateman gradient (*r*)") +
theme_jitter(axis.title.x = element_markdown())
# print
grid.newpage()
pushViewport(viewport(layout = grid.layout(nrow = 1, ncol = 1, widths = unit(1, "npc"))))
print(Plot_AllApproaches1, vp = viewport(layout.pos.row = 1, layout.pos.col = 1))
print(Plot_AllApproaches2, vp = viewport(layout.pos.row = 1, layout.pos.col = 1))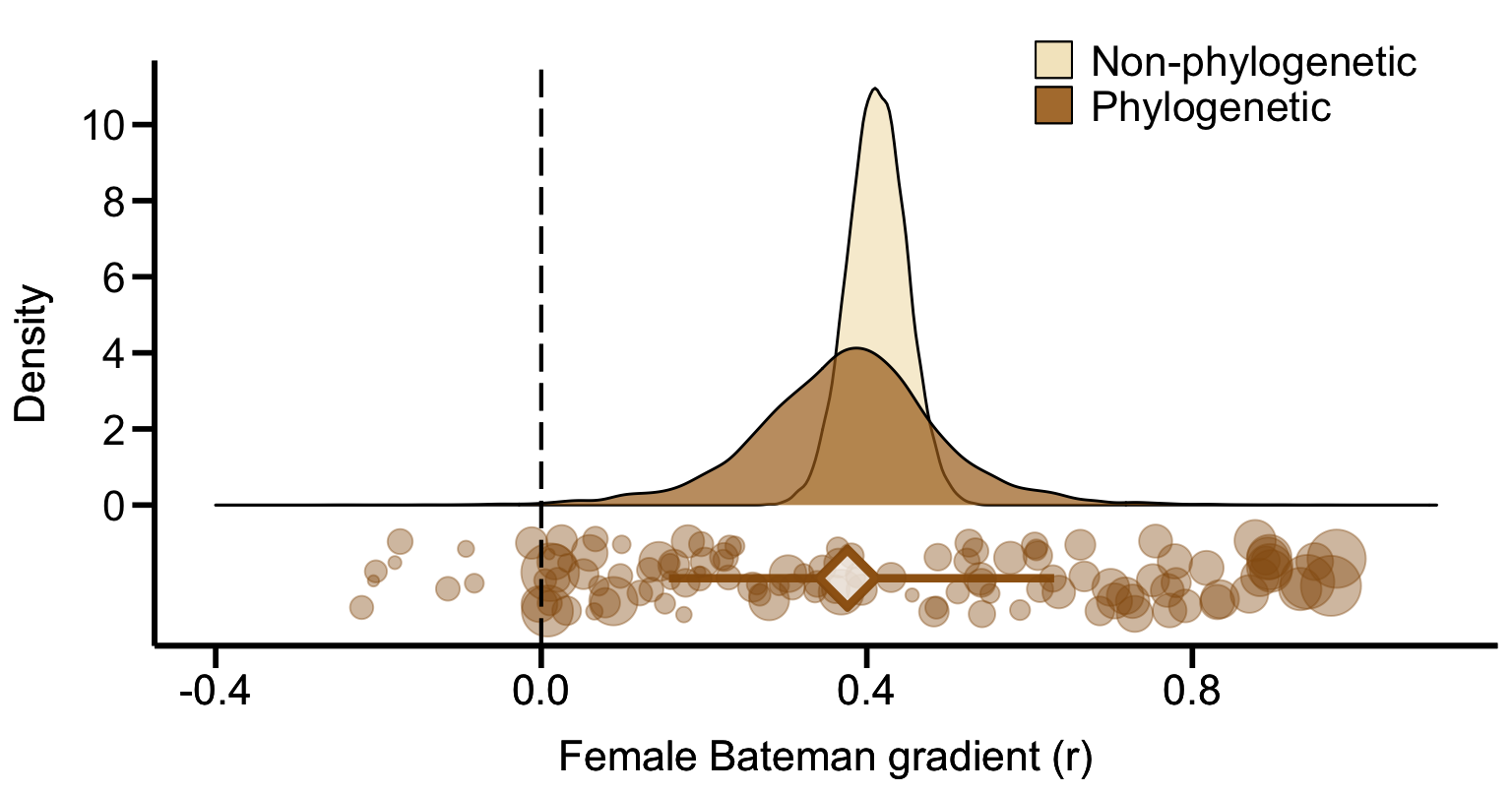
Figure 1A. Meta-analytic evidence for sexual selection in females and its relation to the mating system. Global effect size of the Bateman gradient obtained from Generalized Linear Mixed Models (GLMMs) with or without accounting for phylogenetic non-independence (phylogenetic or non-phylogenetic, respectively). Raincloud charts show posterior distributions, global effect size with 95% Highest Posterior Density intervals (diamonds and error bars) and raw effect sizes (filled circles) of female Bateman gradient.
Moderator variables
Secondly, we ran individual GLMMs in which we defined mating success
method (copulatory versus genetic), mating success range (with versus
without zero-mating success category) or study type (field versus
laboratory studies) as a fixed factor to explain inter-study variation
in r.
These models account for phylogenetic non-independence and include the
phylogeny as a random effect.
Mating success method
MetaData_cMS <- subset(Data, MS_estimate == "cMS")
MetaData_gMS <- subset(Data, MS_estimate == "gMS")
# prune tree ####
MetaData_cMS_Species_Data <- unique(MetaData_cMS$animal)
Tree_cMS <- drop.tip(Tree, Tree$tip.label[-na.omit(match(MetaData_cMS_Species_Data, Tree$tip.label))])
MetaData_gMS_Species_Data <- unique(MetaData_gMS$animal)
Tree_gMS <- drop.tip(Tree, Tree$tip.label[-na.omit(match(MetaData_gMS_Species_Data, Tree$tip.label))])
# predictor model ####
Model_MS_estimate <- MCMCglmm(r_f~factor(MS_estimate),
random = ~animal + Index + Study_ID,
mev = Data$r_VAR_f,
pedigree = Tree,
data = Data,
prior = pr2, pr = TRUE, verbose = FALSE,
burnin = BURNIN, nitt = NITT, thin = THIN)
# null models ####
Model_cMS <- MCMCglmm(r_f~1,
random = ~animal + Index + Study_ID,
mev = MetaData_cMS$r_VAR_f,
pedigree = Tree_cMS,
data = MetaData_cMS,
prior = pr2, pr = TRUE, verbose = FALSE,
burnin = BURNIN, nitt = NITT, thin = THIN)
Model_gMS <- MCMCglmm(r_f~1,
random = ~animal + Index + Study_ID,
mev = MetaData_gMS$r_VAR_f,
pedigree = Tree_gMS,
data = MetaData_gMS,
prior = pr2, pr = TRUE, verbose = FALSE,
burnin = BURNIN, nitt = NITT, thin = THIN)
# Raincloud plot ####
# density
Data_MSest_cMS <- as.data.frame(Model_cMS$Sol[, "(Intercept)"])
Data_MSest_cMS$MatSys <- as.factor(rep("cMS", nrow(Data_MSest_cMS)))
colnames(Data_MSest_cMS) <- c("ES", "MSest")
Data_MSest_gMS <- as.data.frame(Model_gMS$Sol[, "(Intercept)"])
Data_MSest_gMS$MatSys <- as.factor(rep("gMS", nrow(Data_MSest_gMS)))
colnames(Data_MSest_gMS) <- c("ES", "MSest")
Data_MSest_ALL <- as.data.frame(rbind(Data_MSest_cMS, Data_MSest_gMS))
Plot_MSestimate1 <- ggplot() +
geom_density(data = Data_MSest_ALL,
aes(x = ES, y = ..density.., fill = MSest),
alpha = 0.7) +
scale_colour_manual(values = c(TealPalette(20)[9], TealPalette(20)[17])) +
scale_fill_manual(values = c(TealPalette(20)[9], TealPalette(20)[17])) +
geom_vline(xintercept = 0, linetype = "longdash", colour = "black", size = 0.7) +
labs(title = "", x = "", y = "Density") +
scale_x_continuous(limits = c(-0.4, 1.1), breaks = c(-0.4, 0, 0.4, 0.8)) +
scale_y_continuous(limits = c(-3.8, max(density(Data_MSest_cMS$ES)$y)),
breaks = c(0, 2, 4, 6)) +
theme_density(axis.title.x = element_markdown(),
axis.title.y = element_text(size = 22))
# jitter
CI_1 <- HPDinterval(mcmc(Model_cMS$Sol[,"(Intercept)"]))
CI_2 <- HPDinterval(mcmc(Model_gMS$Sol[,"(Intercept)"]))
aggdata_MSest <- data.frame("Jitter" = c("B", "A", "C", "D", "E", "F"),
"MSest" = c("cMS", "gMS", "C", "D", "E", "F"),
"Mean" = c(mean(mcmc(Model_cMS$Sol[, "(Intercept)"])),
mean(mcmc(Model_gMS$Sol[, "(Intercept)"])),
5, 5, 5, 5),
"l025" = c(CI_1[1],
CI_2[1],
5, 5, 5, 5),
"u975" = c(CI_1[2],
CI_2[2],
5, 5, 5, 5))
aggdata_MSest$Jitter <- as.factor(aggdata_MSest$Jitter)
aggdata_MSest$MSest <- as.factor(aggdata_MSest$MSest)
Data$Jitter2 <- as.factor(Data$MS_estimate)
levels(Data$Jitter2) <- c("B", "A")
rm(CI_1, CI_2)
Plot_MSestimate2 <- ggplot() +
geom_pointrange(data = aggdata_MSest,
mapping = aes(x = Mean,
y = Jitter,
xmin = l025,
xmax = u975,
colour = MSest),
shape = 23,
size = 1.5,
fatten = 5,
stroke = 1.75,
fill = "white",
position = position_dodge(width = .7)) +
guides("colour" = "none") +
scale_colour_manual(values = c("black", TealPalette(20)[11], "black", "black", "black", TealPalette(20)[17])) +
new_scale_colour() +
geom_jitter(data = Data,
mapping = aes(x = r_f,
y = Jitter2,
fill = MS_estimate,
colour = MS_estimate,
alpha = MS_estimate),
position = position_jitter(height = 0.35),
size = log(1 / Data$r_VAR_f)) +
scale_alpha_manual(values = c(0.5, 0.4)) +
scale_colour_manual(values = c(TealPalette(20)[11], TealPalette(20)[17])) +
scale_fill_manual(values = c(TealPalette(20)[11], TealPalette(20)[17])) +
new_scale_colour() +
geom_point(data = aggdata_MSest,
mapping = aes(x = Mean,
y = Jitter,
colour = MSest),
shape = 23,
size = 6.5,
stroke = 1.75,
fill = alpha("white", .75),
position = position_dodge(width = .8)) +
guides("colour" = "none") +
geom_vline(xintercept = 0, linetype = "longdash", colour = "black", size = 0.7) +
scale_colour_manual(values = c("black", TealPalette(20)[11], "black", "black", "black", TealPalette(20)[17])) +
scale_x_continuous(limits = c(-0.4, 1.1), breaks = c(-0.4, 0, 0.4, 0.8)) +
scale_y_discrete(labels = c("0", "1", "2", "3", "4", "5")) +
labs(title = "", x = "", y = "Density") +
theme_jitter(axis.title.x = element_markdown(),
axis.title.y = element_text(size = 22))Mating success range
MetaData_with_0MS <- subset(Data, Zero_f == "1")
MetaData_without_0MS <- subset(Data, Zero_f == "0")
# prune tree ####
MetaData_with_0MS_Species_Data <- unique(MetaData_with_0MS$animal)
Tree_with_0MS <- drop.tip(Tree, Tree$tip.label[-na.omit(match(MetaData_with_0MS_Species_Data, Tree$tip.label))])
MetaData_without_0MS_Species_Data <- unique(MetaData_without_0MS$animal)
Tree_without_0MS <- drop.tip(Tree, Tree$tip.label[-na.omit(match(MetaData_without_0MS_Species_Data, Tree$tip.label))])
# predictor model ####
Model_zeroMS <- MCMCglmm(r_f~factor(Zero_f),
random = ~animal + Index + Study_ID,
mev = Data$r_VAR_f,
pedigree = Tree,
data = Data,
prior = pr2, pr = TRUE, verbose = FALSE,
burnin = BURNIN, nitt = NITT, thin = THIN)
# null models ####
Model_with_0MS <- MCMCglmm(r_f~1,
random = ~animal + Index + Study_ID,
mev = MetaData_with_0MS$r_VAR_f,
pedigree = Tree_with_0MS,
data = MetaData_with_0MS,
prior = pr2, pr = TRUE, verbose = FALSE,
burnin = BURNIN, nitt = NITT, thin = THIN)
Model_without_0MS <- MCMCglmm(r_f~1,
random = ~animal + Index + Study_ID,
mev = MetaData_without_0MS$r_VAR_f,
pedigree = Tree_without_0MS,
data = MetaData_without_0MS,
prior = pr2, pr = TRUE, verbose = FALSE,
burnin = BURNIN, nitt = NITT, thin = THIN)
# Raincloud plot ####
# density
Data_ZeroMS_with <- as.data.frame(Model_with_0MS$Sol[, "(Intercept)"])
Data_ZeroMS_with$ZeroMS <- as.factor(rep("With Zero mating success", nrow(Data_ZeroMS_with)))
colnames(Data_ZeroMS_with) <- c("ES", "ZeroMS")
Data_ZeroMS_without <- as.data.frame(Model_without_0MS$Sol[, "(Intercept)"])
Data_ZeroMS_without$ZeroMS <- as.factor(rep("Without Zero mating success", nrow(Data_ZeroMS_without)))
colnames(Data_ZeroMS_without) <- c("ES", "ZeroMS")
Data_ZeroMS_ALL <- as.data.frame(rbind(Data_ZeroMS_with, Data_ZeroMS_without))
Plot_ZeroMS1 <- ggplot() +
geom_density(data = subset(Data_ZeroMS_ALL, Data_ZeroMS_ALL$ZeroMS == "Without Zero mating success"),
aes(x = ES, y = ..density.., fill = "With zero MS"),
alpha = 0.7) +
geom_density(data = subset(Data_ZeroMS_ALL, Data_ZeroMS_ALL$ZeroMS == "With Zero mating success"),
aes(x = ES, y = ..density.., fill = "Without zero MS"),
alpha = 0.7) +
scale_colour_manual(values = c(TealPalette(20)[9], TealPalette(20)[17])) +
scale_fill_manual(labels = c("Without zero MS", "With zero MS"), values = c(TealPalette(20)[9], TealPalette(20)[17])) +
geom_vline(xintercept = 0, linetype = "longdash", colour = "black", size = 0.7) +
labs(title = "", x = "", y = "Density") +
scale_x_continuous(limits = c(-0.4, 1.1), breaks = c(-0.4, 0, 0.4, 0.8)) +
scale_y_continuous(limits = c(-2.8, max(density(Data_ZeroMS_without$ES)$y)),
breaks = c(0, 1, 2, 3, 4, 5)) +
theme_density(axis.title.x = element_markdown(),
axis.title.y = element_text(size = 22))
# jitter
CI_1 <- HPDinterval(mcmc(Model_with_0MS$Sol[,"(Intercept)"]))
CI_2 <- HPDinterval(mcmc(Model_without_0MS$Sol[,"(Intercept)"]))
aggdata_ZeroMS <- data.frame("Jitter" = c("A", "B", "C", "D", "E", "F"),
"ZeroMS" = c("With Zero mating success", "Without Zero mating success", "C", "D", "E", "F"),
"Mean" = c(mean(mcmc(Model_with_0MS$Sol[, "(Intercept)"])),
mean(mcmc(Model_without_0MS$Sol[, "(Intercept)"])),
5, 5, 5, 5),
"l025" = c(CI_1[1],
CI_2[1],
5, 5, 5, 5),
"u975" = c(CI_1[2],
CI_2[2],
5, 5, 5, 5))
aggdata_ZeroMS$Jitter <- as.factor(aggdata_ZeroMS$Jitter)
aggdata_ZeroMS$ZeroMS <- as.factor(aggdata_ZeroMS$ZeroMS)
Data$Jitter2 <- as.factor(Data$Zero_f)
levels(Data$Jitter2) <- c("A", "B")
rm(CI_1, CI_2)
Plot_ZeroMS2 <- ggplot() +
geom_pointrange(data = aggdata_ZeroMS,
mapping = aes(x = Mean,
y = Jitter,
xmin = l025,
xmax = u975,
colour = ZeroMS),
shape = 23,
size = 1.5,
fatten = 5,
stroke = 1.75,
fill = "white",
position = position_dodge(width = .8)) +
guides("colour" = "none") +
scale_colour_manual(values = c("black", "black", "black", "black", TealPalette(20)[17], TealPalette(20)[11])) +
new_scale_colour() +
geom_jitter(data = Data,
mapping = aes(x = r_f,
y = Jitter2,
fill = factor(Zero_f),
colour = factor(Zero_f),
alpha = factor(Zero_f)),
position = position_jitter(height = 0.35),
size = log(1 / Data$r_VAR_f)) +
scale_alpha_manual(values = c(0.4, 0.5)) +
scale_colour_manual(values = c(TealPalette(20)[17], TealPalette(20)[11])) +
scale_fill_manual(values = c(TealPalette(20)[17], TealPalette(20)[11])) +
new_scale_colour() +
geom_point(data = aggdata_ZeroMS,
mapping = aes(x = Mean,
y = Jitter,
colour = ZeroMS),
shape = 23,
size = 6.5,
stroke = 1.75,
fill = alpha("white", .75),
position = position_dodge(width = .7)) +
guides("colour" = "none") +
geom_vline(xintercept = 0, linetype = "longdash", colour = "black", size = 0.7) +
scale_colour_manual(values = c("black", "black", "black", "black", TealPalette(20)[17], TealPalette(20)[11])) +
scale_x_continuous(limits = c(-0.4, 1.1), breaks = c(-0.4, 0, 0.4, 0.8)) +
scale_y_discrete(labels = c("0", "1", "2", "3", "4", "5")) +
labs(title = "", x = "", y = "Density") +
theme_jitter(axis.title.x = element_markdown(),
axis.title.y = element_text(size = 22))Study type
MetaData_Field <- subset(Data, StudyType == "Field")
MetaData_Lab <- subset(Data, StudyType == "Lab")
# prune tree ####
MetaData_Field_Species_Data <- unique(MetaData_Field$animal)
Tree_Field <- drop.tip(Tree, Tree$tip.label[-na.omit(match(MetaData_Field_Species_Data, Tree$tip.label))])
MetaData_Lab_Species_Data <- unique(MetaData_Lab$animal)
Tree_Lab <- drop.tip(Tree, Tree$tip.label[-na.omit(match(MetaData_Lab_Species_Data, Tree$tip.label))])
# predictor model ####
Model_StudyType <- MCMCglmm(r_f~StudyType,
random = ~animal + Index + Study_ID,
mev = Data$r_VAR_f,
pedigree = Tree,
data = Data,
prior = pr2, pr = TRUE, verbose = FALSE,
burnin = BURNIN, nitt = NITT, thin = THIN)
# null models ####
Model_Field <- MCMCglmm(r_f~1,
random = ~animal + Index + Study_ID,
mev = MetaData_Field$r_VAR_f,
pedigree = Tree_Field,
data = MetaData_Field,
prior = pr2, pr = TRUE, verbose = FALSE,
burnin = BURNIN, nitt = NITT, thin = THIN)
Model_Lab <- MCMCglmm(r_f~1,
random = ~animal + Index + Study_ID,
mev = MetaData_Lab$r_VAR_f,
pedigree = Tree_Lab,
data = MetaData_Lab,
prior = pr2, pr = TRUE, verbose = FALSE,
burnin = BURNIN, nitt = NITT, thin = THIN)
# Raincloud plot ####
# density
Data_StudyType_field <- as.data.frame(Model_Field$Sol[, "(Intercept)"])
Data_StudyType_field$StudyType <- as.factor(rep("Field", nrow(Data_StudyType_field)))
colnames(Data_StudyType_field) <- c("ES", "StudyType")
Data_StudyType_lab <- as.data.frame(Model_Lab$Sol[, "(Intercept)"])
Data_StudyType_lab$StudyType <- as.factor(rep("Lab", nrow(Data_StudyType_lab)))
colnames(Data_StudyType_lab) <- c("ES", "StudyType")
Data_StudyType_ALL <- as.data.frame(rbind(Data_StudyType_field, Data_StudyType_lab))
Plot_StudyType1 <- ggplot() +
geom_density(data = Data_StudyType_ALL,
aes(x = ES, y = ..density.., fill = StudyType),
alpha = 0.7) +
scale_colour_manual(values = c(TealPalette(20)[9], TealPalette(20)[17])) +
scale_fill_manual(values = c(TealPalette(20)[9], TealPalette(20)[17])) +
geom_vline(xintercept = 0, linetype = "longdash", colour = "black", size = 0.7) +
labs(title = "", x = "Female Bateman gradient (*r*)", y = "Density") +
scale_x_continuous(limits = c(-0.4, 1.1), breaks = c(-0.4, 0, 0.4, 0.8)) +
scale_y_continuous(limits = c(-2.8, max(density(Data_StudyType_ALL$ES)$y) + 0.7),
breaks = c(0, 1, 2, 3, 4, 5)) +
theme_density(axis.title.x = element_markdown(),
axis.title.y = element_text(size = 22))
# jitter
CI_1 <- HPDinterval(mcmc(Model_Field$Sol[,"(Intercept)"]))
CI_2 <- HPDinterval(mcmc(Model_Lab$Sol[,"(Intercept)"]))
aggdata_StudyType <- data.frame("Jitter" = c("B", "A", "C", "D", "E", "F"),
"StudyType" = c("Field", "Lab", "C", "D", "E", "F"),
"Mean" = c(mean(mcmc(Model_Field$Sol[, "(Intercept)"])),
mean(mcmc(Model_Lab$Sol[, "(Intercept)"])),
5, 5, 5, 5),
"l025" = c(CI_1[1],
CI_2[1],
5, 5, 5, 5),
"u975" = c(CI_1[2],
CI_2[2],
5, 5, 5, 5))
aggdata_StudyType$Jitter <- as.factor(aggdata_StudyType$Jitter)
aggdata_StudyType$StudyType <- as.factor(aggdata_StudyType$StudyType)
Data$Jitter2 <- as.factor(Data$StudyType)
levels(Data$Jitter2) <- c("B", "A")
rm(CI_1, CI_2)
Plot_StudyType2 <- ggplot() +
geom_pointrange(data = aggdata_StudyType,
mapping = aes(x = Mean,
y = Jitter,
xmin = l025,
xmax = u975,
colour = StudyType),
shape = 23,
size = 1.5,
fatten = 5,
stroke = 1.75,
fill = "white",
position = position_dodge(width = .8)) +
guides("colour" = "none") +
scale_colour_manual(values = c("black", "black", "black", "black", TealPalette(20)[11], TealPalette(20)[17])) +
new_scale_colour() +
geom_jitter(data = Data,
mapping = aes(x = r_f,
y = Jitter2,
fill = StudyType,
colour = StudyType,
alpha = StudyType),
position = position_jitter(height = 0.35),
size = log(1 / Data$r_VAR_f)) +
scale_alpha_manual(values = c(0.5, 0.4)) +
scale_colour_manual(values = c(TealPalette(20)[11], TealPalette(20)[17])) +
scale_fill_manual(values = c(TealPalette(20)[11], TealPalette(20)[17])) +
new_scale_colour() +
geom_point(data = aggdata_StudyType,
mapping = aes(x = Mean,
y = Jitter,
colour = StudyType),
shape = 23,
size = 6.5,
stroke = 1.75,
fill = alpha("white", .75),
position = position_dodge(width = .7)) +
guides("colour" = "none") +
geom_vline(xintercept = 0, linetype = "longdash", colour = "black", size = 0.7) +
scale_colour_manual(values = c("black", "black", "black", "black", TealPalette(20)[11], TealPalette(20)[17])) +
scale_x_continuous(limits = c(-0.4, 1.1), breaks = c(-0.4, 0, 0.4, 0.8)) +
scale_y_discrete(labels = c("0", "1", "2", "3", "4", "5")) +
labs(title = "", x = "Female Bateman gradient (*r*)", y = "Density") +
theme_jitter(axis.title.x = element_markdown(),
axis.title.y = element_text(size = 22))Plot_Density2 <- plot_grid(Plot_MSestimate1, Plot_ZeroMS1, Plot_StudyType1,
labels = "AUTO",
label_size = 18,
hjust = 0,
vjust = 1,
align = "hv",
ncol = 1,
nrow = 3)
Plot_Jitter2 <- plot_grid(Plot_MSestimate2, Plot_ZeroMS2, Plot_StudyType2,
labels = "AUTO",
label_size = 18,
hjust = 0,
vjust = 1,
align = "hv",
ncol = 1,
nrow = 3)
grid.newpage()
pushViewport(viewport(layout = grid.layout(nrow = 1, ncol = 1, widths = unit(1, "npc"))))
print(Plot_Density2, vp = viewport(layout.pos.row = 1, layout.pos.col = 1))
print(Plot_Jitter2, vp = viewport(layout.pos.row = 1, layout.pos.col = 1))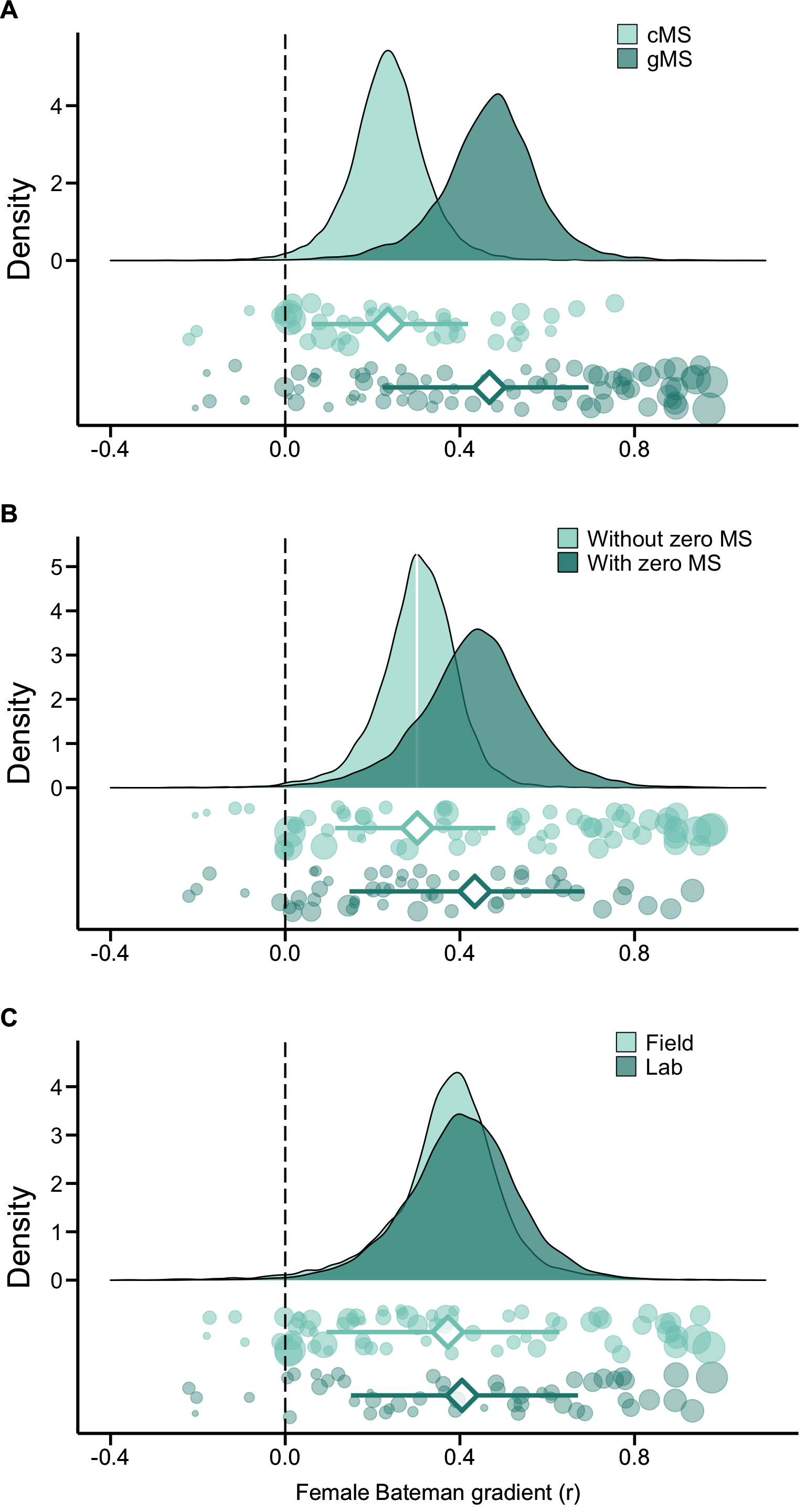
Figure S2. Methodological predictors of female Bateman gradients. Raincloud charts showing effects of mating success method (cMS: copulatory mating success, gMS: genetic mating success), mating success range (with or without zero mating success (MS) category) and study type (field versus laboratory studies) on female Bateman gradients (for statistical analysis see Table 2 and S3).
Mating system
We classified the mating system of each sampled species based on estimates of polyandry, which we defined as the proportion of reproducing females that have more than one mating partner. We then used these estimates to define the mating system as either monandrous or polyandrous, depending on whether its value was lower or higher than 0.5, respectively.
MetaData_polygamous <- subset(Data, MatingSystem == "high polyandry")
MetaData_monogamous <- subset(Data, MatingSystem == "low polyandry")
# prune tree ####
MetaData_polygamous_Species_Data <- unique(MetaData_polygamous$animal)
Tree_polygamous <- drop.tip(Tree, Tree$tip.label[-na.omit(match(MetaData_polygamous_Species_Data, Tree$tip.label))])
MetaData_monogamous_Species_Data <- unique(MetaData_monogamous$animal)
Tree_monogamous <- drop.tip(Tree, Tree$tip.label[-na.omit(match(MetaData_monogamous_Species_Data, Tree$tip.label))])
# predictor model ####
Model_MatingSystem <- MCMCglmm(r_f~MatingSystem,
random = ~animal + Index + Study_ID,
mev = Data$r_VAR_f,
pedigree = Tree,
data = Data,
prior = pr2, pr = TRUE, verbose = FALSE,
burnin = BURNIN, nitt = NITT, thin = THIN)
# null models ####
Model_monogamous <- MCMCglmm(r_f~1,
random = ~animal + Index + Study_ID,
mev = MetaData_monogamous$r_VAR_f,
pedigree = Tree_monogamous,
data = MetaData_monogamous,
prior = pr2, pr = TRUE, verbose = FALSE,
burnin = BURNIN, nitt = NITT, thin = THIN)
Model_polygamous <- MCMCglmm(r_f~1,
random = ~animal + Index + Study_ID,
mev = MetaData_polygamous$r_VAR_f,
pedigree = Tree_polygamous,
data = MetaData_polygamous,
prior = pr2, pr = TRUE, verbose = FALSE,
burnin = BURNIN, nitt = NITT, thin = THIN)
# Raincloud plot ####
# density
Data_MatSys_Monogamy <- as.data.frame(Model_monogamous$Sol[, "(Intercept)"])
Data_MatSys_Monogamy$MatSys <- as.factor(rep("Low polyandry", nrow(Data_MatSys_Monogamy)))
colnames(Data_MatSys_Monogamy) <- c("ES", "MatSys")
Data_MatSys_Polygamy <- as.data.frame(Model_polygamous$Sol[, "(Intercept)"])
Data_MatSys_Polygamy$MatSys <- as.factor(rep("High polyandry", nrow(Data_MatSys_Polygamy)))
colnames(Data_MatSys_Polygamy) <- c("ES", "MatSys")
Data_MatingSystem_ALL <- as.data.frame(rbind(Data_MatSys_Monogamy, Data_MatSys_Polygamy))
Plot_MatingSystem1 <- ggplot() +
geom_density(data = Data_MatingSystem_ALL,
aes(x = ES, y = ..density.., fill = MatSys),
alpha = 0.7) +
scale_colour_manual(values = c(TealPalette(20)[9], TealPalette(20)[17])) +
scale_fill_manual(values = c(TealPalette(20)[9], TealPalette(20)[17])) +
geom_vline(xintercept = 0, linetype = "longdash", colour = "black", size = 0.7) +
labs(title = "", x = "Female Bateman gradient (*r*)", y = "Density") +
scale_x_continuous(limits = c(-0.4, 1.1), breaks = c(-0.4, 0, 0.4, 0.8)) +
scale_y_continuous(limits = c(-2.5, 5), breaks = c(0, 1, 2, 3, 4, 5)) +
theme_density(axis.title.x = element_markdown())
# jitter
CI_1 <- HPDinterval(mcmc(Model_monogamous$Sol[,"(Intercept)"]))
CI_2 <- HPDinterval(mcmc(Model_polygamous$Sol[,"(Intercept)"]))
aggdata_MatSys <- data.frame("Jitter" = c("A", "B", "C", "D", "E", "F"),
"MatSys" = c("High polyandry", "Low polyandry", "C", "D", "E", "F"),
"Mean" = c(mean(mcmc(Model_polygamous$Sol[, "(Intercept)"])),
mean(mcmc(Model_monogamous$Sol[, "(Intercept)"])),
5, 5, 5, 5),
"l025" = c(CI_2[1],
CI_1[1],
5, 5, 5, 5),
"u975" = c(CI_2[2],
CI_1[2],
5, 5, 5, 5))
aggdata_MatSys$Jitter <- as.factor(aggdata_MatSys$Jitter)
aggdata_MatSys$MatSys <- as.factor(aggdata_MatSys$MatSys)
Data$Jitter2 <- as.factor(Data$MatingSystem)
levels(Data$Jitter2) <- c("B", "A")
rm(CI_1, CI_2)
Plot_MatingSystem2 <- ggplot() +
geom_pointrange(data = aggdata_MatSys,
mapping = aes(x = Mean,
y = Jitter,
xmin = l025,
xmax = u975,
colour = MatSys),
shape = 23,
size = 1.5,
fatten = 5,
stroke = 1.75,
fill = "white",
position = position_dodge(width = .8)) +
scale_colour_manual(values = c("black", "black", "black", "black", TealPalette(20)[17], TealPalette(20)[11])) +
new_scale_colour() +
geom_jitter(data = Data,
mapping = aes(x = r_f,
y = Jitter2,
fill = MatingSystem,
colour = MatingSystem,
alpha = MatingSystem),
position = position_jitter(height = 0.35),
size = log(1 / Data$r_VAR_f)) +
scale_colour_manual(values = c(TealPalette(20)[11], TealPalette(20)[17])) +
scale_fill_manual(values = c(TealPalette(20)[11], TealPalette(20)[17])) +
scale_alpha_manual(values = c(0.5, 0.4)) +
new_scale_colour() +
geom_point(data = aggdata_MatSys,
mapping = aes(x = Mean,
y = Jitter,
colour = MatSys),
shape = 23,
size = 6.5,
stroke = 1.75,
fill = alpha("white", .75),
position = position_dodge(width = .8)) +
geom_vline(xintercept = 0, linetype = "longdash", colour = "black", size = 0.7) +
scale_colour_manual(values = c("black", "black", "black", "black", TealPalette(20)[17], TealPalette(20)[11])) +
scale_x_continuous(limits = c(-0.4, 1.1), breaks = c(-0.4, 0, 0.4, 0.8)) +
scale_y_discrete(labels = c("0", "1", "2", "3", "4", "5")) +
labs(title = "", x = "Female Bateman gradient (*r*)", y = "Density") +
theme_jitter(axis.title.x = element_markdown())
# print
grid.newpage()
pushViewport(viewport(layout = grid.layout(nrow = 1, ncol = 1, widths = unit(1, "npc"))))
print(Plot_MatingSystem1, vp = viewport(layout.pos.row = 1, layout.pos.col = 1))
print(Plot_MatingSystem2, vp = viewport(layout.pos.row = 1, layout.pos.col = 1))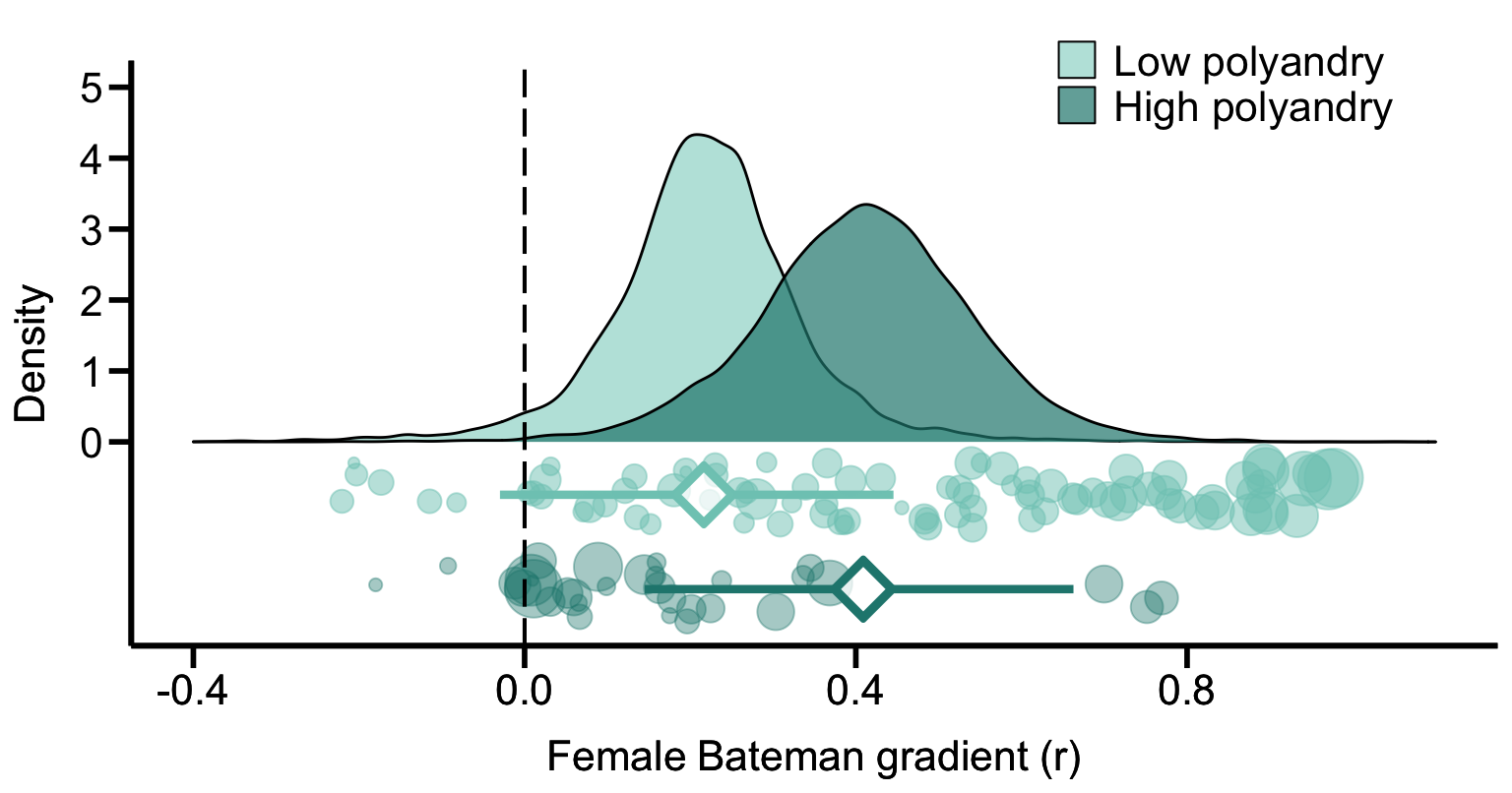
Figure 1B. Meta-analytic evidence for sexual selection in females and its relation to the mating system. Contrast in sexual selection in females between low-polyandry and high-polyandry species. Raincloud charts show posterior distributions, global effect size with 95% Highest Posterior Density intervals (diamonds and error bars) and raw effect sizes (filled circles) of female Bateman gradient.
Polyandry
We also ran GLMMs with the proportion of multiply mated females (i.
e., a continuous variable) as a fixed effect.
The studentized Breusch-Pagan test informs on whether this variable is
significantly heteroscedastic.
# predictor model ####
Model_Polyandry <- MCMCglmm(r_f ~ Polyandry,
random = ~animal + Index + Study_ID,
mev = Data$r_VAR_f,
pedigree = Tree,
data = Data,
prior = pr2, pr = TRUE, verbose = FALSE,
burnin = BURNIN, nitt = NITT, thin = THIN)
## The Breusch-Pagan Test ####
LM_Polyandry_r_f <- lm(r_f ~ Polyandry, data = Data)
intercept <- summary(Model_Polyandry)$solutions[1, 1]
slope <- summary(Model_Polyandry)$solutions[2, 1]
slope_l95 <- summary(Model_Polyandry)$solutions[2, 2]
slope_u95 <- summary(Model_Polyandry)$solutions[2, 3]
pointsize_r_f <- log(1/Data$r_VAR_f)
Data$Polyandry_residuals <- LM_Polyandry_r_f$residuals
LM_BreuschPagan_Polyandry <- lm(Polyandry_residuals^2 ~ Polyandry, data = Data)
summary(LM_BreuschPagan_Polyandry)
Call:
lm(formula = Polyandry_residuals^2 ~ Polyandry, data = Data)
Residuals:
Min 1Q Median 3Q Max
-0.13438 -0.07606 -0.02805 0.03455 0.44389
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 0.02617 0.02515 1.040 0.30025
Polyandry 0.11042 0.03862 2.859 0.00502 **
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.1093 on 118 degrees of freedom
Multiple R-squared: 0.06479, Adjusted R-squared: 0.05687
F-statistic: 8.175 on 1 and 118 DF, p-value: 0.005022# plot ####
Plot_r_f_Polyandry <- ggplot(Data, aes(x = Polyandry, y = r_f)) +
geom_point(shape = 19, size = pointsize_r_f, alpha = 0.6, colour = "royalblue") +
labs(title = "", x = "Polyandry", y = "Bateman gradient (*r*)") +
scale_x_continuous(limits = c(-0.1, 1.1),
breaks = c(0, 0.25, 0.5, 0.75, 1),
expand = c(0, 0)) +
scale_y_continuous(limits = c(-0.5, 1.2),
breaks = c(-0.5, 0, 0.5, 1),
expand = c(0, 0)) +
theme_paper(axis.title.y = element_markdown()) +
geom_abline(aes(intercept = intercept, slope = slope), colour = "black")
Plot_r_f_Polyandry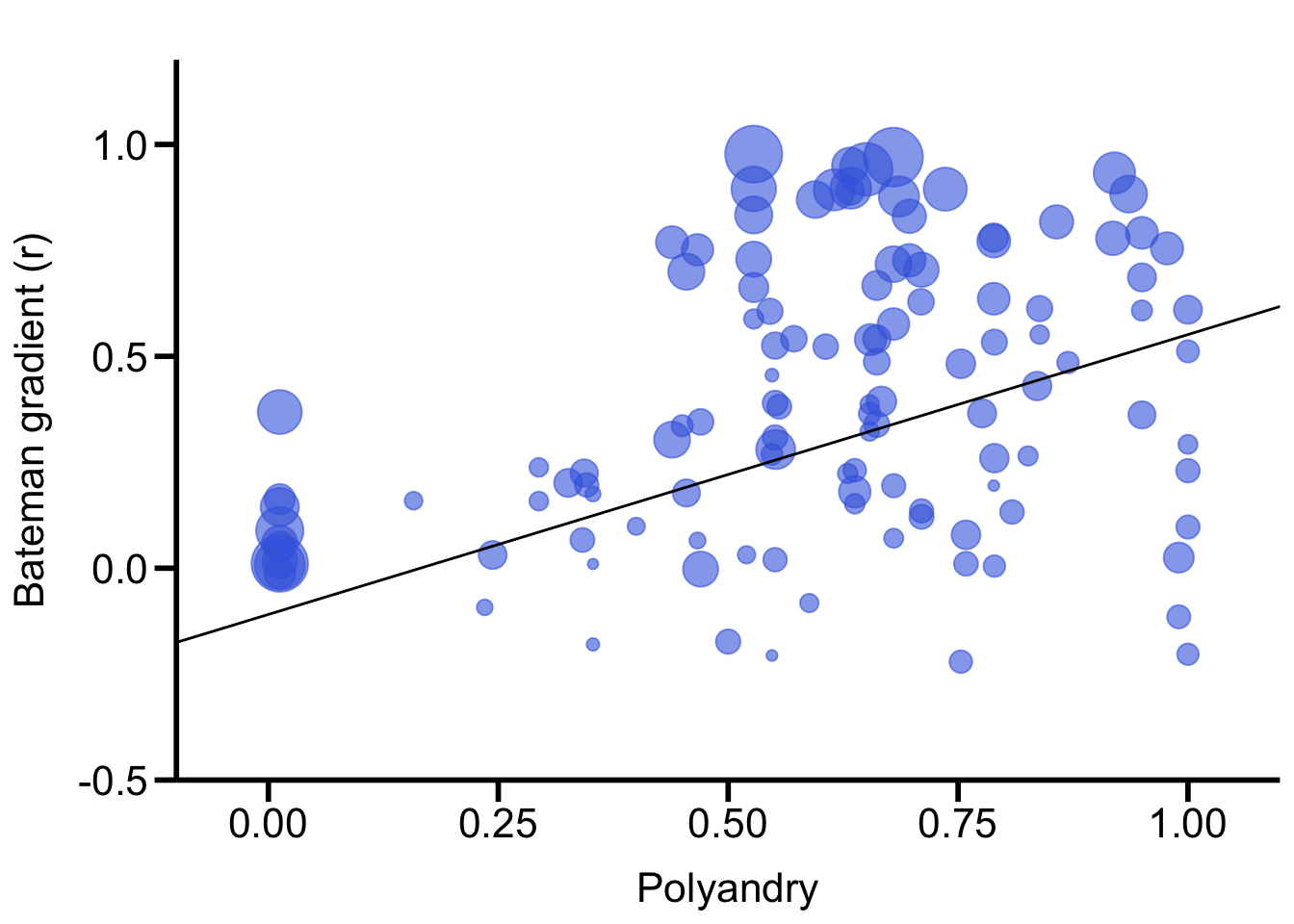
Publication year
We tested for the effect of the publication year of the studies on the effect size to investigate the presence or absence of the bandwagon effect.
# predictor model
Model_Year <- MCMCglmm(r_f~Year,
random = ~animal + Index + Study_ID,
mev = Data$r_VAR_f,
pedigree = Tree,
data = Data,
prior = pr2, pr = TRUE, verbose = FALSE,
burnin = BURNIN, nitt = NITT, thin = THIN)Heterogeneity
We created functions to extract the relevant information from the models and to estimate heterogeneity I², which we defined as the proportion of variance in effect size that can be attributed to the different levels of random effects (Higgins and Thompson 2002). In particular, we decomposed total heterogeneity into the proportional phylogenetic variance (I²Phylogeny), between-study variance (I²Study), and study-specific variance (observation-level random effect; I²Observation) (Nakagawa and Santos 2012). For models including predictor variables, we computed the proportion of variance explained by those fixed factors (‘marginal R²’) (Nakagawa and Schielzeth 2013).
# Null models ####
NullModel_summary1 <- function(which_data) {
k <- length(which_data$r_f) # sample size
N_sp <- length((aggregate(r_f ~ animal, data = which_data, FUN = sum))$animal) # number of species
as.data.frame(cbind(k, N_sp))
}
NullModel_summary2 <- function(which_model) {
# MCMCglmm summary
MCMCglmm_summary <- as.data.frame(cbind(
posterior.mode(which_model$Sol[,"(Intercept)"]), # posterior mode
cbind(HPDinterval(which_model$Sol[,"(Intercept)"])), # 95% highest posterior density
summary(which_model)$solutions[, 5])) #P(MCMC)
colnames(MCMCglmm_summary) <- c("Mode", "Lower HPD", "Higher HPD", "pmcmc")
# heterogeneity
## I2 Phylogenetic signal - Mode and HPD
H2_null_models <- which_model$VCV[, "animal"]/(which_model$VCV[, "animal"] + which_model$VCV[, "Study_ID"] + which_model$VCV[, "Index"] + which_model$VCV[, "units"])
Mode_H2 <- posterior.mode(H2_null_models)
HPD_H2 <- HPDinterval(H2_null_models)
ALL_H2 <- as.data.frame(cbind(Mode_H2, HPD_H2))
colnames(ALL_H2) <- c("Mode2", "Lower HPD2", "Higher HPD2")
## I2 Study ID variance - Mode and HPD
Study2_null_models <- which_model$VCV[, "Study_ID"]/(which_model$VCV[, "animal"] + which_model$VCV[, "Study_ID"] + which_model$VCV[, "Index"] + which_model$VCV[, "units"])
Mode_Study2 <- posterior.mode(Study2_null_models)
HPD_Study2 <- HPDinterval(Study2_null_models)
ALL_Study2 <- as.data.frame(cbind(Mode_Study2, HPD_Study2))
colnames(ALL_Study2) <- c("Mode3", "Lower HPD3", "Higher HPD3")
## I2 Observation ID variance - Mode and HPD
Index2_null_models <- which_model$VCV[, "Index"]/(which_model$VCV[, "animal"] + which_model$VCV[, "Study_ID"] + which_model$VCV[, "Index"] + which_model$VCV[, "units"])
Mode_Index2 <- posterior.mode(Index2_null_models)
HPD_Index2 <- HPDinterval(Index2_null_models)
ALL_Index2 <- as.data.frame(cbind(Mode_Index2, HPD_Index2))
colnames(ALL_Index2) <- c("Mode4", "Lower HPD4", "Higher HPD4")
# output
as.data.frame(cbind(MCMCglmm_summary,
ALL_H2,
ALL_Study2,
ALL_Index2))
}
List_data <- list(Data,
MetaData_cMS, MetaData_gMS,
MetaData_with_0MS, MetaData_without_0MS,
MetaData_Lab, MetaData_Field,
MetaData_monogamous, MetaData_polygamous)
List_Nullmodels <- list(Model_ALL_phylo,
Model_cMS, Model_gMS,
Model_with_0MS, Model_without_0MS,
Model_Lab, Model_Field,
Model_monogamous, Model_polygamous)
Model_sum1 <- adply(.data = List_data, .margins = 1, .fun = NullModel_summary1) # result of function as a dataframe: each row is f(x) for a different x from the list
Model_sum2 <- adply(.data = List_Nullmodels, .margins = 1, .fun = NullModel_summary2)
Table1 <- cbind(Model_sum1[,-1], Model_sum2[, -1]) %>%
mutate_at(c(3:5, 7:15), round, digits = 2)
Table1 <- Table1 %>%
mutate_at(6, round, digits = 3)
# MCMC model summary - traditional approach
MCMCglmm_Model_ALL_traditional <- as.data.frame(cbind(
posterior.mode(Model_ALL_traditional$Sol[,"(Intercept)"]),
cbind(HPDinterval(Model_ALL_traditional$Sol[,"(Intercept)"])),
summary(Model_ALL_traditional)$solutions[, 5]))
colnames(MCMCglmm_Model_ALL_traditional) <- c("Mode", "Lower HPD", "Higher HPD", "pmcmc")
MCMCglmm_Model_ALL_traditional_sum <- cbind(k <- length(Data$r_f),
N_sp <- length((aggregate(r_f ~ animal, data = Data, FUN = sum))$animal),
MCMCglmm_Model_ALL_traditional,
t(rep(c("--", "", ""), times = 3)))
colnames(MCMCglmm_Model_ALL_traditional_sum) <- c("k", "N_sp", "Mode", "Lower HPD", "Higher HPD", "pmcmc", "Mode2", "Lower HPD2", "Higher HPD2", "Mode3", "Lower HPD3", "Higher HPD3", "Mode4", "Lower HPD4", "Higher HPD4")
MCMCglmm_Model_ALL_traditional_sum[, 3:5] <- round(MCMCglmm_Model_ALL_traditional_sum[, 3:5], digits = 2)
MCMCglmm_Model_ALL_traditional_sum[, 6] <- round(MCMCglmm_Model_ALL_traditional_sum[, 6], digits = 3)
# Predictor models ####
PredictorModel_summary <- function(which_model) {
# MCMCglmm summary
MCMCglmm_summary <- as.data.frame(cbind(
posterior.mode(which_model$Sol[, 2]), # posterior mode
cbind(HPDinterval(which_model$Sol[, 2])), # 95% highest posterior density
summary(which_model)$solutions[2, 5])) #P(MCMC)
colnames(MCMCglmm_summary) <- c("Mode", "Lower HPD", "Higher HPD", "pmcmc")
# Explained variance
mPred_Model <- mean(which_model$Sol[, 2]) * which_model$X[, 2]
mVar_mPred_Model <- var(mPred_Model)
R2_Pred_models <- mVar_mPred_Model/(mVar_mPred_Model + which_model$VCV[, "animal"] + which_model$VCV[, "Study_ID"] + which_model$VCV[, "Index"] + which_model$VCV[, "units"])
Mode_R2 <- posterior.mode(R2_Pred_models)
HPD_R2 <- HPDinterval(R2_Pred_models)
ALL_R2 <- as.data.frame(cbind(Mode_R2, HPD_R2))
colnames(ALL_R2) <- c("R2 Mode", "R2 Lower HPD", "R2 Higher HPD")
as.data.frame(cbind(MCMCglmm_summary, ALL_R2))
}
List_Predmodels <- list(Model_MS_estimate,
Model_zeroMS,
Model_StudyType,
Model_Year,
Model_MatingSystem,
Model_Polyandry)
Table2 <- adply(.data = List_Predmodels, .margins = 1, .fun = PredictorModel_summary) %>%
select(2:8) %>%
mutate_at(c(1:3, 5:7), round, digits = 2)
Table2 <- Table2 %>%
mutate_at(4, round, digits = 3)Models summary
Table 1. Global tests of sexual selection in females. Results of intercept-only phylogenetically controlled General Linear-Mixed Effects Models are shown for the entire dataset (global model) and subsets with respect to mating success method (copulatory versus genetic), mating success range (including versus excluding zero mating success category), study type (laboratory versus field studies) and mating system (low-polyandry versus high-polyandry species). Table shows number of effect sizes (k), number of species (N), effect size (r), and heterogeneity I² arising from phylogenetic affinities, between-study variation, and between-observation variation. Model estimates are shown as posterior modes with 95% Highest Posterior Density (HPD) intervals in brackets.
for (i in c(4, 8, 11, 14)){
Table1[, i] <- paste0("(", Table1[, i], ", ")}
for (i in c(5, 9, 12, 15)){
Table1[, i] <- paste0(Table1[, i], ")")}
MCMCglmm_Model_ALL_traditional_sum$`Lower HPD` <- paste0("(", MCMCglmm_Model_ALL_traditional_sum$`Lower HPD`, ", ")
MCMCglmm_Model_ALL_traditional_sum$`Higher HPD` <- paste0(MCMCglmm_Model_ALL_traditional_sum$`Higher HPD`, ")")
# All null models summary
Table1 <- rbind(MCMCglmm_Model_ALL_traditional_sum, Table1)
colnames(Table1) <- c("", "", "r", "", "", "P~MCMC~",
"I²~Phylogeny~", "", "",
"I²~Study~", "", "",
"I²~Observation~", "", "")
rownames(Table1) <- c("Global model (non-phylogenetic)",
"Global model (phylogenetic)",
"Copulatory mating success",
"Genetic mating success",
"Including zero mating success",
"Excluding zero mating success",
"Laboratory studies",
"Field studies",
"Low-polyandry species",
"High-polyandry species")
Table1[1, 6] <- "<0.001"
Table1 %>%
kable(align = c(rep("c", 3), "r", "l", "r", rep(c("c", "r", "l"), 3))) %>%
kable_paper(full_width = TRUE) %>%
add_header_above(c("Model" = 1, "k" = 1, "N~Species~" = 1, "Effect size" = 4, "Heterogeneity" = 9)) %>%
row_spec(0, italic = TRUE, font_size = 14)|
Model
|
k
|
NSpecies
|
Effect size
|
Heterogeneity
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| r | PMCMC | I²Phylogeny | I²Study | I²Observation | |||||||||||
| Global model (non-phylogenetic) | 120 | 77 | 0.42 | (0.35, | 0.49) | <0.001 | – | – | – | ||||||
| Global model (phylogenetic) | 120 | 77 | 0.38 | (0.16, | 0.63) | 0.006 | 0.04 | (0.01, | 0.8) | 0.26 | (0.04, | 0.78) | 0.01 | (0, | 0.25) |
| Copulatory mating success | 43 | 24 | 0.23 | (0.06, | 0.42) | 0.02 | 0.06 | (0.01, | 0.79) | 0.02 | (0, | 0.46) | 0.02 | (0, | 0.64) |
| Genetic mating success | 77 | 54 | 0.49 | (0.22, | 0.69) | 0.003 | 0.01 | (0, | 0.74) | 0.66 | (0.11, | 0.9) | 0.01 | (0, | 0.24) |
| Including zero mating success | 66 | 41 | 0.44 | (0.15, | 0.69) | 0.007 | 0.68 | (0.01, | 0.86) | 0.01 | (0, | 0.68) | 0.01 | (0, | 0.36) |
| Excluding zero mating success | 54 | 42 | 0.30 | (0.11, | 0.48) | 0.011 | 0.01 | (0, | 0.61) | 0.8 | (0.19, | 0.94) | 0.02 | (0, | 0.33) |
| Laboratory studies | 52 | 31 | 0.39 | (0.15, | 0.67) | 0.009 | 0.79 | (0.11, | 0.92) | 0.01 | (0, | 0.51) | 0.01 | (0, | 0.41) |
| Field studies | 68 | 47 | 0.40 | (0.09, | 0.63) | 0.026 | 0.01 | (0, | 0.81) | 0.84 | (0.1, | 0.94) | 0.01 | (0, | 0.19) |
| Low-polyandry species | 32 | 16 | 0.20 | (-0.03, | 0.45) | 0.071 | 0.02 | (0, | 0.68) | 0.8 | (0.01, | 0.9) | 0.01 | (0, | 0.49) |
| High-polyandry species | 88 | 61 | 0.41 | (0.15, | 0.66) | 0.006 | 0.74 | (0.29, | 0.92) | 0.01 | (0, | 0.41) | 0.01 | (0, | 0.31) |
Table 2. Predictors of inter-specific variation in
female Bateman gradients. Methodological moderators include
mating success method (copulatory versus genetic mating success), mating
success range (including versus excluding zero mating success category),
study type (field versus lab) and year of publication (continuous
variable). Effect of mating system contrasts low-polyandry and
high-polyandry species. Effect of polyandry estimates the relationship
between the female Bateman gradient and the proportion of polyandrous
females in the population. Model estimates (i.e., estimated difference
between groups) are shown as posterior modes with 95% Highest Posterior
Density (HPD) intervals obtained from phylogenetically controlled
General Linear-Mixed Effects Models. The variance explained by the
moderator variable is given as the marginal R² with 95% HPD intervals in
brackets.
for (i in c(2, 6)){
Table2[, i] <- paste0("(", Table2[, i], ", ")}
for (i in c(3, 7)){
Table2[, i] <- paste0(Table2[, i], ")")}
# All predictor models summary
rownames(Table2) <- c("Mating success method",
"Mating success range",
"Study type",
"Year of publication",
"Mating system",
"Polyandry")
#Table2[1, 6] <- "<0.001"
Table2 %>%
kable(align = c("c", "r", "l", "r", "c", "r", "l"),
col.names = c("Estimate", "", "", "P~MCMC~",
"R²", "", "")) %>%
kable_paper(full_width = FALSE)| Estimate | PMCMC | R² | |||||
|---|---|---|---|---|---|---|---|
| Mating success method | 0.28 | (0.11, | 0.45) | 0.001 | 0.17 | (0.09, | 0.22) |
| Mating success range | 0.16 | (0.03, | 0.28) | 0.015 | 0.04 | (0.02, | 0.07) |
| Study type | 0.08 | (-0.11, | 0.25) | 0.475 | 0.01 | (0, | 0.01) |
| Year of publication | -0.01 | (-0.02, | 0.01) | 0.422 | 0.01 | (0, | 0.01) |
| Mating system | -0.33 | (-0.47, | -0.16) | 0.000 | 0.14 | (0.08, | 0.22) |
| Polyandry | 0.67 | (0.38, | 0.94) | 0.000 | 0.16 | (0.1, | 0.27) |
Restricted Maximum Likelihood Meta-Analysis
For completeness, we also ran all GLMMs using the Restricted Maximum
Likelihood (REML) approach using the metafor package (Viechtbauer 2010).
Data <- read.table("./data/Fromonteil_et_al_DATA.txt", fileEncoding = "UTF-8") # the file encoding supports special characters
Tree <- read.tree("./data/Fromonteil_et_al_TREE.txt")
MetaData_All_Species_Data <- unique(Data$Species_Phylo)
Tree <- drop.tip(Tree, Tree$tip.label[-na.omit(match(MetaData_All_Species_Data, Tree$tip.label))])
# Run Models ####
# All
forcedC_ALL <- as.matrix(forceSymmetric(vcv(Tree, corr = TRUE))) # formatting for the phylogenetic tree
Model_REML_ALL <- rma.mv(r_f ~ 1,
V = r_VAR_f,
data = Data,
random = c(~ 1 | Study_ID, ~ 1 | Index, ~ 1 | Species_Phylo),
R = list(Species_Phylo = forcedC_ALL), method = "REML")
# Mating success method
forcedC_cMS <- as.matrix(forceSymmetric(vcv(Tree_cMS, corr = TRUE)))
forcedC_gMS <- as.matrix(forceSymmetric(vcv(Tree_gMS, corr = TRUE)))
MetaData_cMS <- subset(Data, MS_estimate == "cMS")
MetaData_gMS <- subset(Data, MS_estimate == "gMS")
Model_REML_MS_Method <- rma.mv(r_f ~ factor(MS_estimate),
V = r_VAR_f,
data = Data,
random = c(~ 1 | Study_ID, ~ 1 | Index, ~ 1 | Species_Phylo),
R = list(Species_Phylo = forcedC_ALL), method = "REML")
Model_REML_cMS <- rma.mv(r_f ~ 1,
V = r_VAR_f,
data = MetaData_cMS,
random = c(~ 1 | Study_ID, ~ 1 | Index, ~ 1 | Species_Phylo),
R = list(Species_Phylo = forcedC_cMS), method = "REML")
Model_REML_gMS <- rma.mv(r_f ~ 1,
V = r_VAR_f,
data = MetaData_gMS,
random = c(~ 1 | Study_ID, ~ 1 | Index, ~ 1 | Species_Phylo),
R = list(Species_Phylo = forcedC_gMS), method = "REML")
# Mating success range
forcedC_with_0MS <- as.matrix(forceSymmetric(vcv(Tree_with_0MS, corr = TRUE)))
forcedC_without_0MS <- as.matrix(forceSymmetric(vcv(Tree_without_0MS, corr = TRUE)))
MetaData_with_0MS <- subset(Data, Zero_f == "1")
MetaData_without_0MS <- subset(Data, Zero_f == "0")
Model_REML_MS_Zero <- rma.mv(r_f ~ factor(Zero_f),
V = r_VAR_f,
data = Data,
random = c(~ 1 | Study_ID, ~ 1 | Index, ~ 1 | Species_Phylo),
R = list(Species_Phylo = forcedC_ALL), method = "REML")
Model_REML_with_0MS <- rma.mv(r_f ~ 1,
V = r_VAR_f,
data = MetaData_with_0MS,
random = c(~ 1 | Study_ID, ~ 1 | Index, ~ 1 | Species_Phylo),
R = list(Species_Phylo = forcedC_with_0MS), method = "REML")
Model_REML_without_0MS = rma.mv(r_f ~ 1,
V = r_VAR_f,
data = MetaData_without_0MS,
random = c(~ 1 | Study_ID, ~ 1 | Index, ~ 1 | Species_Phylo),
R = list(Species_Phylo = forcedC_without_0MS), method = "REML")
# Study type
forcedC_Field <- as.matrix(forceSymmetric(vcv(Tree_Field, corr = TRUE)))
forcedC_Lab <- as.matrix(forceSymmetric(vcv(Tree_Lab, corr = TRUE)))
MetaData_Field <- subset(Data, StudyType == "Field")
MetaData_Lab <- subset(Data, StudyType == "Lab")
Model_REML_StudyType <- rma.mv(r_f ~ factor(StudyType),
V = r_VAR_f,
data = Data,
random = c(~ 1 | Study_ID, ~ 1 | Index, ~ 1 | Species_Phylo),
R = list(Species_Phylo = forcedC_ALL), method = "REML")
Model_REML_Field <- rma.mv(r_f ~ 1,
V = r_VAR_f,
data = MetaData_Field,
random = c(~ 1 | Study_ID, ~ 1 | Index, ~ 1 | Species_Phylo),
R = list(Species_Phylo = forcedC_Field), method = "REML")
Model_REML_Lab <- rma.mv(r_f ~ 1,
V = r_VAR_f,
data = MetaData_Lab,
random = c(~ 1 | Study_ID, ~ 1 | Index, ~ 1 | Species_Phylo),
R = list(Species_Phylo = forcedC_Lab), method = "REML")
# Mating system
forcedC_polygamous <- as.matrix(forceSymmetric(vcv(Tree_polygamous, corr = TRUE)))
forcedC_monogamous <- as.matrix(forceSymmetric(vcv(Tree_monogamous, corr = TRUE)))
MetaData_polygamous <- subset(Data, MatingSystem == "high polyandry")
MetaData_monogamous <- subset(Data, MatingSystem == "low polyandry")
Model_REML_MatingSystem <- rma.mv(r_f ~ factor(MatingSystem),
V = r_VAR_f,
data = Data,
random = c(~ 1 | Study_ID, ~ 1 | Index, ~ 1 | Species_Phylo),
R = list(Species_Phylo = forcedC_ALL), method = "REML")
Model_REML_polygamous <- rma.mv(r_f ~ 1,
V = r_VAR_f,
data = MetaData_polygamous,
random = c(~ 1 | Study_ID, ~ 1 | Index, ~ 1 | Species_Phylo),
R = list(Species_Phylo = forcedC_polygamous), method = "REML")
Model_REML_monogamous <- rma.mv(r_f ~ 1,
V = r_VAR_f,
data = MetaData_monogamous,
random = c(~ 1 | Study_ID, ~ 1 | Index, ~ 1 | Species_Phylo),
R = list(Species_Phylo = forcedC_monogamous), method = "REML")
# Polyandry
Model_REML_Polyandry <- rma.mv(r_f ~ Polyandry,
V = r_VAR_f,
data = Data,
random = c(~ 1 | Study_ID, ~ 1 | Index, ~ 1 | Species_Phylo),
R = list(Species_Phylo = forcedC_ALL), method = "REML")
# Year
Model_REML_Year <- rma.mv(r_f ~ Year,
V = r_VAR_f,
data = Data,
random = c(~ 1 | Study_ID, ~ 1 | Index, ~ 1 | Species_Phylo),
R = list(Species_Phylo = forcedC_ALL), method = "REML")Models summary
Table S1. Global tests of sexual selection in females using the restricted maximum likelihood (REML) approach. Results of intercept-only phylogenetically controlled General Linear-Mixed Effects Models are shown for the entire dataset (global model) and subsets with respect to mating success method (copulatory versus genetic), mating success range (including versus excluding zero mating success category), study type (laboratory versus field studies) and mating system (low-polyandry versus high-polyandry species). Table shows number of effect sizes (k), number of species (N) and estimates of r together with 95% confidence intervals (in brackets).
REMLNullModel_summary1 <- function(which_data) {
k <- length(which_data$r_f) # sample size
N_sp <- length((aggregate(r_f ~ Species_Phylo, data = which_data, FUN = sum))$Species_Phylo) # number of species
as.data.frame(cbind(k, N_sp))
}
REMLNullModel_summary2 <- function(which_model) {
# REML summary
as.data.frame(cbind(
summary(which_model)$"beta", # global effect size
summary(which_model)$ci.lb, # 95% confidence interval - low
summary(which_model)$ci.ub, # 95% confidence interval - high
summary(which_model)$zval, # Z-value
summary(which_model)$pval)) # P-value
}
List_data <- list(Data,
MetaData_cMS, MetaData_gMS,
MetaData_with_0MS, MetaData_without_0MS,
MetaData_Lab, MetaData_Field,
MetaData_monogamous, MetaData_polygamous)
List_Nullmodels <- list(Model_REML_ALL,
Model_REML_cMS, Model_REML_gMS,
Model_REML_with_0MS, Model_REML_without_0MS,
Model_REML_Lab, Model_REML_Field,
Model_REML_monogamous, Model_REML_polygamous)
Model_sum1 <- adply(.data = List_data, .margins = 1, .fun = REMLNullModel_summary1)
Model_sum2 <- adply(.data = List_Nullmodels, .margins = 1, .fun = REMLNullModel_summary2)
TableS1 <- cbind(Model_sum1[,-1], Model_sum2[, -1]) %>%
mutate_at(c(3:5), round, digits = 2)
TableS1 <- TableS1 %>%
mutate_at(6:7, round, digits = 3)
for (i in c(4)){
TableS1[, i] <- paste0("(", TableS1[, i], ", ")}
for (i in c(5)){
TableS1[, i] <- paste0(TableS1[, i], ")")}
colnames(TableS1) <- c("", "", "r", "", "", "z-value", "P-value")
rownames(TableS1) <- c("Global model",
"Copulatory mating success",
"Genetic mating success",
"Including zero mating success",
"Excluding zero mating success",
"Laboratory studies",
"Field studies",
"Low-polyandry species",
"High-polyandry species")
#TableS1[1, 6] <- "<0.001"
TableS1 %>%
kable(align = c(rep("c", 3), "r", "l", "c", "c")) %>%
kable_paper(full_width = T) %>%
add_header_above(c("Model" = 1, "k" = 1, "N~Species~" = 1, "Global effect size" = 3, " " = 2)) %>%
row_spec(0, italic = T, font_size = 14)|
Model
|
k
|
NSpecies
|
Global effect size
|
||||
|---|---|---|---|---|---|---|---|
| r | z-value | P-value | |||||
| Global model | 120 | 77 | 0.37 | (0.15, | 0.59) | 3.329 | 0.001 |
| Copulatory mating success | 43 | 24 | 0.23 | (0.06, | 0.41) | 2.655 | 0.008 |
| Genetic mating success | 77 | 54 | 0.47 | (0.26, | 0.67) | 4.437 | 0.000 |
| Including zero mating success | 66 | 41 | 0.43 | (0.17, | 0.69) | 3.262 | 0.001 |
| Excluding zero mating success | 54 | 42 | 0.33 | (0.23, | 0.42) | 6.679 | 0.000 |
| Laboratory studies | 52 | 31 | 0.40 | (0.15, | 0.65) | 3.104 | 0.002 |
| Field studies | 68 | 47 | 0.40 | (0.31, | 0.49) | 8.963 | 0.000 |
| Low-polyandry species | 32 | 16 | 0.23 | (0.09, | 0.37) | 3.161 | 0.002 |
| High-polyandry species | 88 | 61 | 0.41 | (0.16, | 0.66) | 3.202 | 0.001 |
Table S3. Predictors of inter-specific variation in
female Bateman gradients using restricted maximum likelihood (REML)
approach. Methodological moderators include mating success
method (copulatory versus genetic mating success), mating success range
(including versus excluding mating success category), study type (field
versus lab) and year of publication (continuous variable). Effect of
mating system contrasts low-polyandry and high-polyandry species. Effect
of polyandry (continuous variable) estimates the relationship between
the female Bateman gradient and the proportion of polyandrous females in
the population. Phylogenetically controlled multilevel meta-analytic
single predictor models are shown with Omnibus tests (Wald-type
chi-square test) and McFadden’s R².
REMLPredictorModel_summary <- function(which_model) {
# REML summary
as.data.frame(cbind(
coef(summary(which_model))[2, 1], # global effect size
coef(summary(which_model))[2, 2], # standard error
summary(which_model)$"QM", # QM: Omnibus test
summary(which_model)$"QMp", # P-value of QM
1 - (logLik.rma(which_model)/logLik.rma(Model_REML_ALL)))) # R²: McFadden's R2
}
List_Predmodels <- list(Model_REML_MS_Method,
Model_REML_MS_Zero,
Model_REML_StudyType,
Model_REML_Year,
Model_REML_MatingSystem,
Model_REML_Polyandry)
Model_sum <- adply(List_Predmodels, 1, REMLPredictorModel_summary)
TableS2 <- cbind(Model_sum[,-1]) %>%
mutate_at(c(1:3), round, digits = 2)
TableS2 <- TableS2 %>%
mutate_at(4, round, digits = 3)
for (i in c(2)){
TableS2[, i] <- paste0("±", TableS2[, i])}
colnames(TableS2) <- c("Estimate", "± SE", "Q~M~", "P-value", "R²")
rownames(TableS2) <- c("Mating success method",
"Mating success range",
"Study type",
"Year",
"Mating system",
"Polyandry")
#TableS1[1, 6] <- "<0.001"
TableS2 %>%
kable(align = c("r", "l", "c", "c")) %>%
kable_paper(full_width = F)| Estimate | ± SE | QM | P-value | R² | |
|---|---|---|---|---|---|
| Mating success method | 0.28 | ±0.09 | 10.71 | 0.001 | 0.2303087 |
| Mating success range | 0.16 | ±0.06 | 6.48 | 0.011 | 0.1415606 |
| Study type | 0.07 | ±0.09 | 0.62 | 0.429 | 0.0258698 |
| Year | 0.00 | ±0.01 | 0.67 | 0.414 | 0.0128597 |
| Mating system | -0.31 | ±0.07 | 16.92 | 0.000 | 0.3518470 |
| Polyandry | 0.66 | ±0.14 | 23.30 | 0.000 | 0.4717672 |
Publication bias
Lastly, we build funnel plots to visually assess asymmetry.
# Funnel plot for raw Bateman gradients
# Define model
forcedC <- as.matrix(forceSymmetric(vcv(Tree, corr = TRUE)))
Model_REML_ALL_by_Moderator <- rma.mv(r_f ~ factor(MatingSystem) + factor(MS_estimate) + factor(Zero_f) + factor(StudyType) + Year,
V = r_VAR_f,
data = Data,
random = c(~ 1 | Study_ID, ~ 1 | Index, ~ 1 | Species_Phylo),
R = list(Species_Phylo = forcedC), method = "REML")
Data$Residuals_Phylo <- resid(Model_REML_ALL)
Data$Residuals_Phylo_Moderator <- resid(Model_REML_ALL_by_Moderator)
# extract relevant information
Data$r_SE <- sqrt(Data$r_VAR_f) # standard error
effect_size_mode <- posterior.mode(Model_ALL_phylo$Sol[, "(Intercept)"]) # mode
effect_size_ll95 <- summary(Model_ALL_phylo)$solutions[, 2] # lower 95% highest posterior density
effect_size_ul95 <- summary(Model_ALL_phylo)$solutions[, 3] # upper 95% highest posterior density
# Additional lines on funnel plot
se.seq <- seq(0, max(Data$r_SE), 0.001) # range from 0 to the max SE in the data
# 95% confidence interval
ll95 <- effect_size_mode - (1.96*se.seq)
ul95 <- effect_size_mode + (1.96*se.seq)
# 99% confidence interval
ll99 <- effect_size_mode - (3.29*se.seq)
ul99 <- effect_size_mode + (3.29*se.seq)
dfCI <- data.frame(ll95, ul95,
ll99, ul99,
se.seq, effect_size_mode,
effect_size_ll95, effect_size_ul95)
# plot
FunnelPlot <- ggplot() +
geom_line(data = dfCI,
mapping = aes(x = se.seq, y = ll95),
linetype = "solid", colour = "#B2D8D8", size = 1) +
geom_line(data = dfCI,
mapping = aes(x = se.seq, y = ul95),
linetype = "solid", colour = "#B2D8D8", size = 1) +
geom_line(data = dfCI,
mapping = aes(x = se.seq, y = ll99),
linetype = "solid", colour = "#008080", size = 1) +
geom_line(data = dfCI,
mapping = aes(x = se.seq, y = ul99),
linetype = "solid", colour = "#008080", size = 1) +
geom_segment(data = dfCI,
mapping = aes(x = min(se.seq), y = effect_size_mode, xend = max(se.seq), yend = effect_size_mode),
linetype = "dashed", size = 0.7) +
geom_point(data = Data,
mapping = aes(x = r_SE, y = r_f),
shape = 21, size = 4, fill = "grey", alpha = .6) +
labs(title = "", x = "Standard error", y = "Bateman gradient (*r*)") +
scale_y_continuous(breaks = c(-1.5, 2, 0.5),
labels = scales::number_format(accuracy = 0.1),
limits = c(-1.5, 2)) +
scale_x_reverse() +
coord_flip() +
theme_paper(axis.title.x = element_markdown())
# Funnel plot for residual Bateman Gradients
### Customized Egger's regression ###
Data$Precision <- 1/(Data$r_SE)
Data$SND_NullModel <- Data$Residuals_Phylo/(Data$r_SE)
Data$SND_ModeratorModel <- Data$Residuals_Phylo_Moderator/(Data$r_SE)
estimate_RES <- mean(Data$Residuals_Phylo_Moderator)
se <- sd(Data$Residuals_Phylo_Moderator)/sqrt(length(Data$Residuals_Phylo_Moderator))
# Additional lines on res funnel plot
# 95% confidence interval
Res_ll95 = 0 - (1.96*se.seq)
Res_ul95 = 0 + (1.96*se.seq)
# 99% confidence interval
Res_ll99 = 0 - (3.29*se.seq)
Res_ul99 = 0 + (3.29*se.seq)
Res_dfCI <- data.frame(Res_ll95, Res_ul95,
Res_ll99, Res_ul99,
se.seq, estimate_RES)
# plot
FunnelPlot_Resid <- ggplot() +
geom_line(data = Res_dfCI,
mapping = aes(x = se.seq, y = Res_ll95),
linetype = "solid", colour = "#B2D8D8", size = 1) +
geom_line(data = Res_dfCI,
mapping = aes(x = se.seq, y = Res_ul95),
linetype = "solid", colour = "#B2D8D8", size = 1) +
geom_line(data = Res_dfCI,
mapping = aes(x = se.seq, y = Res_ll99),
linetype = "solid", colour = "#008080", size = 1) +
geom_line(data = Res_dfCI,
mapping = aes(x = se.seq, y = Res_ul99),
linetype = "solid", colour = "#008080", size = 1) +
geom_segment(data = Res_dfCI,
mapping = aes(x = min(se.seq), y = 0, xend = max(se.seq), yend = 0),
linetype = "dashed", size = 0.7) +
geom_point(data = Data,
mapping = aes(x = r_SE, y = Residuals_Phylo_Moderator),
shape = 21, size = 4, fill = "grey", alpha = .6) +
labs(title = "", x = "Standard error", y = "Residual Bateman gradient (*r*)") +
scale_y_continuous(breaks = c(-1.5, 2, 0.5),
labels = scales::number_format(accuracy = 0.1),
limits = c(-1.5, 2)) +
scale_x_reverse() +
coord_flip() +
theme_paper(axis.title.x = element_markdown())
Figure_FunnelPlots <- plot_grid(FunnelPlot, FunnelPlot_Resid,
labels = "AUTO",
label_size = 15,
hjust = 0,
vjust = 1,
align = "hv",
ncol = 1,
nrow = 2)
Figure_FunnelPlots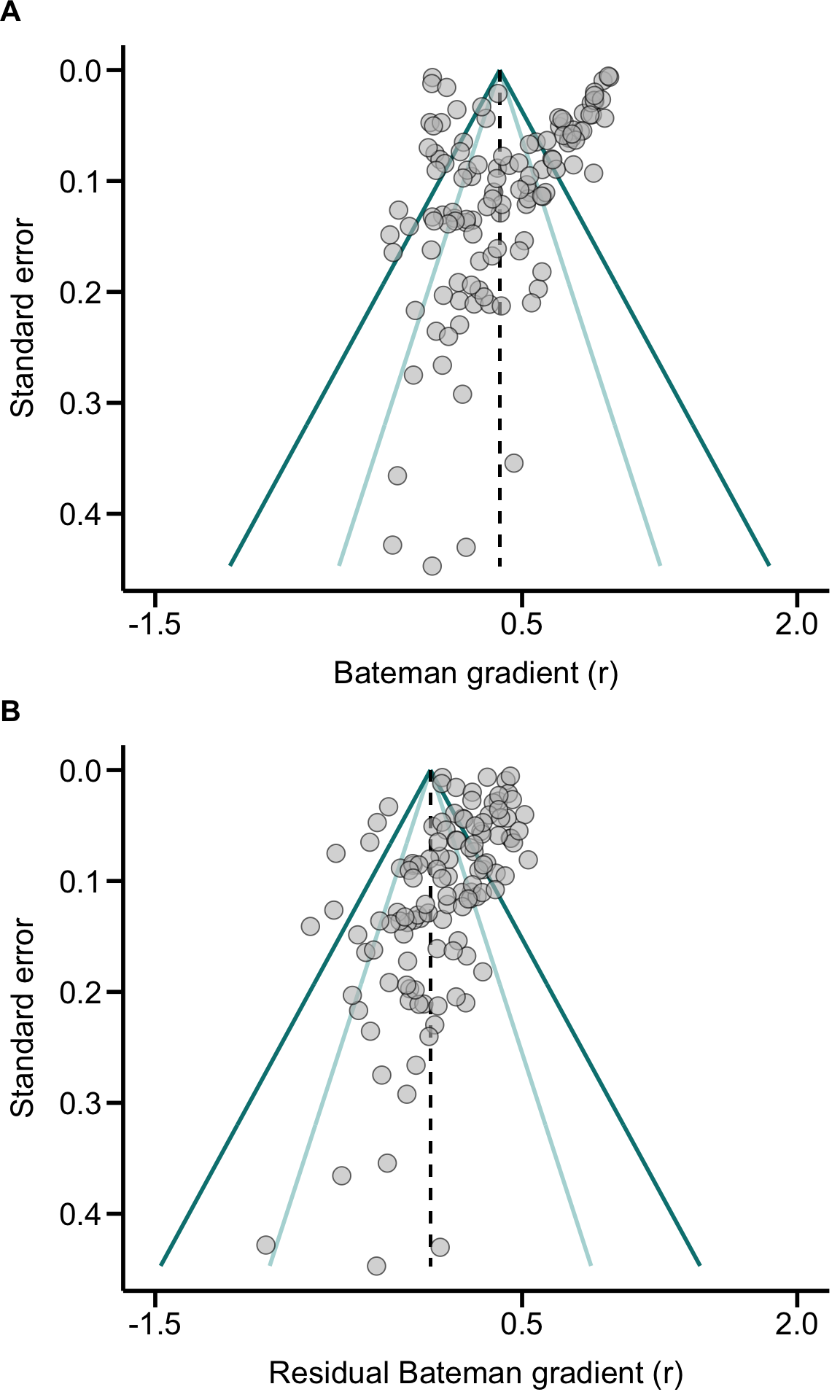
Figure S3. Funnel plots. Data are shown for (A) raw values and (B) for meta-analytic residuals obtained from multivariate linear mixed-effects models accounting for all tested moderator variables. Dashed lines indicate the estimated global effect size. Dark and light blue solid lines denote the expected 95% and 99% confidence limits purely due to sampling heterogeneity. Asymmetries along the global effect size may reflect publication biases.
References
sessionInfo()R version 4.2.0 (2022-04-22)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur/Monterey 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] grid stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] knitr_1.39 htmlTable_2.4.1 kableExtra_1.3.4 ggtext_0.1.1
[5] ggnewscale_0.4.7 RColorBrewer_1.1-3 ggtree_3.4.0 data.table_1.14.2
[9] dplyr_1.0.9 plyr_1.8.7 cowplot_1.1.1 ggplot2_3.3.6
[13] MCMCglmm_2.34 coda_0.19-4 ape_5.6-2 metafor_3.4-0
[17] metadat_1.2-0 Matrix_1.4-1 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] nlme_3.1-158 fs_1.5.2 webshot_0.5.3 httr_1.4.3
[5] rprojroot_2.0.3 tensorA_0.36.2 tools_4.2.0 backports_1.4.1
[9] bslib_0.4.0 utf8_1.2.2 R6_2.5.1 mgcv_1.8-40
[13] lazyeval_0.2.2 colorspace_2.0-3 withr_2.5.0 tidyselect_1.1.2
[17] processx_3.7.0 compiler_4.2.0 git2r_0.30.1 cli_3.3.0
[21] rvest_1.0.2 xml2_1.3.3 labeling_0.4.2 sass_0.4.2
[25] scales_1.2.0 checkmate_2.1.0 callr_3.7.1 systemfonts_1.0.4
[29] stringr_1.4.0 digest_0.6.29 yulab.utils_0.0.5 rmarkdown_2.14
[33] svglite_2.1.0 pkgconfig_2.0.3 htmltools_0.5.3 fastmap_1.1.0
[37] highr_0.9 htmlwidgets_1.5.4 rlang_1.0.4 rstudioapi_0.13
[41] farver_2.1.1 gridGraphics_0.5-1 jquerylib_0.1.4 generics_0.1.3
[45] jsonlite_1.8.0 magrittr_2.0.3 ggplotify_0.1.0 patchwork_1.1.1
[49] Rcpp_1.0.9 munsell_0.5.0 fansi_1.0.3 lifecycle_1.0.1
[53] stringi_1.7.8 whisker_0.4 yaml_2.3.5 mathjaxr_1.6-0
[57] parallel_4.2.0 promises_1.2.0.1 crayon_1.5.1 lattice_0.20-45
[61] splines_4.2.0 gridtext_0.1.4 ps_1.7.1 pillar_1.8.0
[65] cubature_2.0.4.4 markdown_1.1 corpcor_1.6.10 glue_1.6.2
[69] evaluate_0.15 getPass_0.2-2 ggfun_0.0.6 vctrs_0.4.1
[73] treeio_1.20.0 httpuv_1.6.5 gtable_0.3.0 purrr_0.3.4
[77] tidyr_1.2.0 cachem_1.0.6 xfun_0.31 tidytree_0.3.9
[81] later_1.3.0 viridisLite_0.4.0 tibble_3.1.8 aplot_0.1.6
[85] ellipsis_0.3.2